Departament de Genètica Animal, Centre de Recerca en Agrigenòmica, CRAG, Universitat Autònoma de Barcelona, Barcelona, Spain.
PLoS One. 2012;7(4):e35349. doi: 10.1371/journal.pone.0035349. Epub 2012 Apr 25.
The current disease model for leishmaniasis suggests that only a proportion of infected individuals develop clinical disease, while others are asymptomatically infected due to immune control of infection. The factors that determine whether individuals progress to clinical disease following Leishmania infection are unclear, although previous studies suggest a role for host genetics. Our hypothesis was that canine leishmaniasis is a complex disease with multiple loci responsible for the progression of the disease from Leishmania infection.
METHODOLOGY/PRINCIPAL FINDINGS: Genome-wide association and genomic selection approaches were applied to a population-based case-control dataset of 219 dogs from a single breed (Boxer) genotyped for ~170,000 SNPs. Firstly, we aimed to identify individual disease loci; secondly, we quantified the genetic component of the observed phenotypic variance; and thirdly, we tested whether genome-wide SNP data could accurately predict the disease.
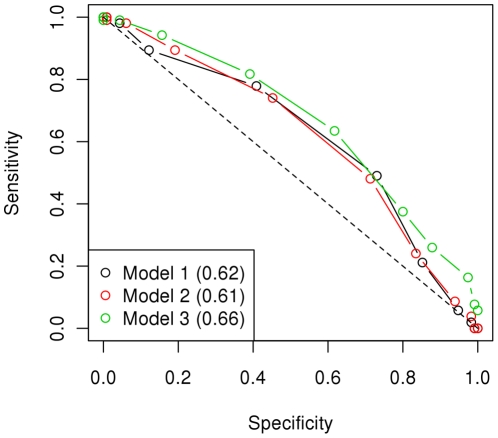
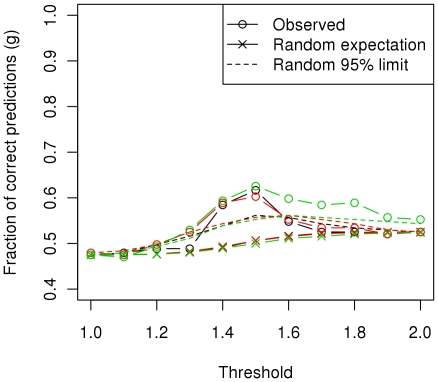
CONCLUSIONS/SIGNIFICANCE: We estimated that a substantial proportion of the genome is affecting the trait and that its heritability could be as high as 60%. Using the genome-wide association approach, the strongest associations were on chromosomes 1, 4 and 20, although none of these were statistically significant at a genome-wide level and after correcting for genetic stratification and lifestyle. Amongst these associations, chromosome 4: 61.2-76.9 Mb maps to a locus that has previously been associated with host susceptibility to human and murine leishmaniasis, and genomic selection estimated markers in this region to have the greatest effect on the phenotype. We therefore propose these regions as candidates for replication studies. An important finding of this study was the significant predictive value from using the genomic information. We found that the phenotype could be predicted with an accuracy of ~0.29 in new samples and that the affection status was correctly predicted in 60% of dogs, significantly higher than expected by chance, and with satisfactory sensitivity-specificity values (AUC = 0.63).
目前的利什曼病发病模型表明,只有一部分感染者会发展为临床疾病,而其他人则由于对感染的免疫控制而无症状感染。在利什曼原虫感染后,决定个体是否进展为临床疾病的因素尚不清楚,尽管先前的研究表明宿主遗传学可能起作用。我们的假设是,犬利什曼病是一种复杂的疾病,多个基因座负责从利什曼原虫感染向疾病的发展。
方法/主要发现:我们对来自单一品种(拳师犬)的 219 只犬进行了基于群体的病例对照数据集,对其进行了全基因组关联和基因组选择分析,这些犬被基因分型为约 170000 个 SNP。首先,我们旨在确定个体疾病基因座;其次,我们量化了观察到的表型方差的遗传成分;第三,我们测试了全基因组 SNP 数据是否可以准确预测疾病。
结论/意义:我们估计,基因组的很大一部分都在影响着这一特征,其遗传率可能高达 60%。使用全基因组关联方法,最强的关联位于第 1、4 和 20 号染色体上,但这些染色体在全基因组水平上均无统计学意义,且在纠正遗传分层和生活方式的影响后仍无统计学意义。在这些关联中,第 4 号染色体:61.2-76.9 Mb 映射到一个先前与人类和鼠利什曼病宿主易感性相关的基因座,基因组选择估计该区域的标记对表型有最大的影响。因此,我们将这些区域作为候选基因进行复制研究。本研究的一个重要发现是使用基因组信息具有显著的预测价值。我们发现,在新样本中,表型可以预测的准确率约为 0.29,在 60%的犬中,疾病状态的预测是正确的,这明显高于预期的随机水平,且具有令人满意的敏感性-特异性值(AUC = 0.63)。