Zalpuri Saurabh, Zwaginga Jaap Jan, van der Bom J G
Sanquin-LUMC Jon J van Rood Center for Clinical Transfusion Research, Leiden, the Netherlands.
BMJ Open. 2012 May 4;2(3). doi: 10.1136/bmjopen-2012-001150. Print 2012.
Individuals exposed to red blood cell alloantigens through transfusion, pregnancy or transplantation may produce antibodies against the alloantigens. Alloantibodies can pose serious clinical problems such as delayed haemolytic reactions and logistic problems, for example, to obtain timely and properly matched transfusion blood for patients in which new alloantibodies are detected.
The authors hypothesise that the particular clinical conditions (eg, used medication, concomitant infection, cellular immunity) during which transfusions are given may contribute to the risk of immunisation. The aim of this research was to examine the association between clinical, environmental and genetic characteristics of the recipient of erythrocyte transfusions and the risk against erythrocyte alloimmunisation during that transfusion episode. METHODS AND ANALYSIS STUDY DESIGN: Incident case-cohort study.
Secondary care, nationwide study (within the Netherlands) including seven hospitals, from January 2005 to December 2011.
Consecutive red cell transfused patients at the study centres.
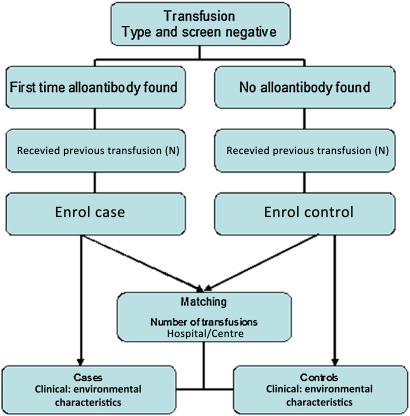
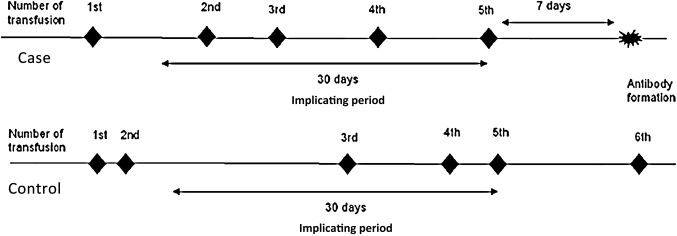
The study cohort comprises of consecutive red blood cell transfused patients at the study centre. EXCLUSION: Patients with transfusions before the study period and/or pre-existing alloantibodies.Cases defined as first time alloantibody formers; Controls defined as transfused individuals matched (on number of transfusions) to cases and have not formed an alloantibody.
Logistic regression models will be used to assess the association between the risk to develop antibodies and potential risk factors, adjusted for other risk factors.
Approval at each local ethics regulatory committee will be obtained. Data will be coded for privacy reasons. Patients will be sent a letter and an information brochure explaining the purpose of the study. A consent form in presence of the study coordinator will be signed before the blood taking commences. Investigators will submit progress summary of the study to study sponsor regularly. Investigators will notify the accredited ethics board of the end of the study within a period of 8 weeks.
通过输血、妊娠或移植接触红细胞同种异体抗原的个体可能会产生针对这些同种异体抗原的抗体。同种抗体可能会引发严重的临床问题,如迟发性溶血反应,以及后勤问题,例如,为检测到新同种抗体的患者及时获取匹配得当的输血用血。
作者推测输血时的特定临床状况(如所用药物、合并感染、细胞免疫)可能会增加免疫风险。本研究的目的是探讨红细胞输血受者的临床、环境和遗传特征与该次输血期间红细胞同种免疫风险之间的关联。
方法与分析
病例队列研究。
二级医疗,全国性研究(在荷兰境内),涵盖七家医院,时间跨度为2005年1月至2011年12月。
研究中心连续接受红细胞输血的患者。
研究队列包括研究中心连续接受红细胞输血的患者。排除标准:研究期间之前接受过输血和/或已有同种抗体的患者。病例定义为首次产生同种抗体者;对照定义为与病例(按输血量)匹配且未产生同种抗体的输血个体。
将使用逻辑回归模型评估产生抗体的风险与潜在风险因素之间未经调整和经其他风险因素调整后的关联。
将获得各地方伦理监管委员会的批准。出于隐私原因,数据将进行编码。将向患者发送一封信件和一份信息手册,解释研究目的。在采血开始前,将在研究协调员在场的情况下签署同意书。研究人员将定期向研究资助者提交研究进展摘要。研究人员将在8周内通知认可的伦理委员会研究结束。