Department of Neurosciences NC30, Lerner Research Institute, The Cleveland Clinic Foundation, 9500 Euclid Avenue, Cleveland, OH 44195, USA.
J Neuroinflammation. 2012 May 29;9:104. doi: 10.1186/1742-2094-9-104.
The interplay between IFN-γ, IL-17 and neutrophils during CNS inflammatory disease is complex due to cross-regulatory factors affecting both positive and negative feedback loops. These interactions have hindered the ability to distinguish the relative contributions of neutrophils, Th1 and Th17 cell-derived effector molecules from secondary mediators to tissue damage and morbidity.
Encephalitis induced by a gliatropic murine coronavirus was used as a model to assess the direct contributions of neutrophils, IFN-γ and IL-17 to virus-induced mortality. CNS inflammatory conditions were selectively manipulated by adoptive transfer of virus-primed wild-type (WT) or IFN-γ deficient (GKO) memory CD4+ T cells into infected SCID mice, coupled with antibody-mediated neutrophil depletion and cytokine blockade.
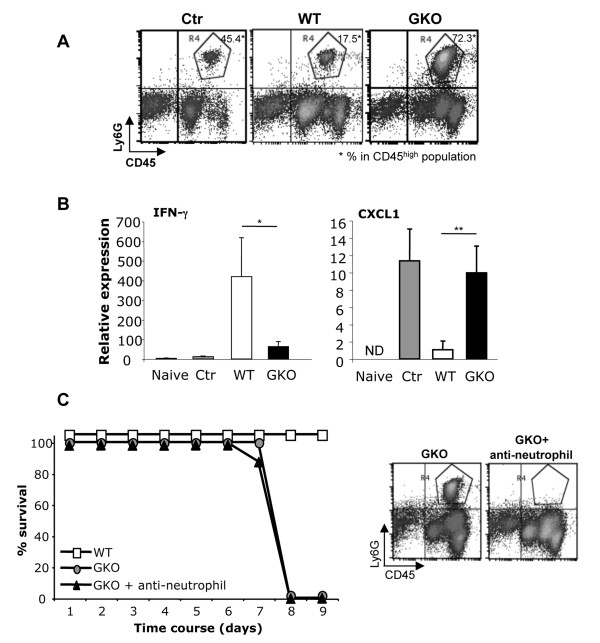
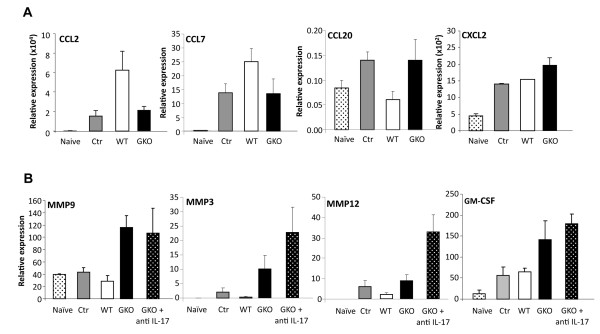
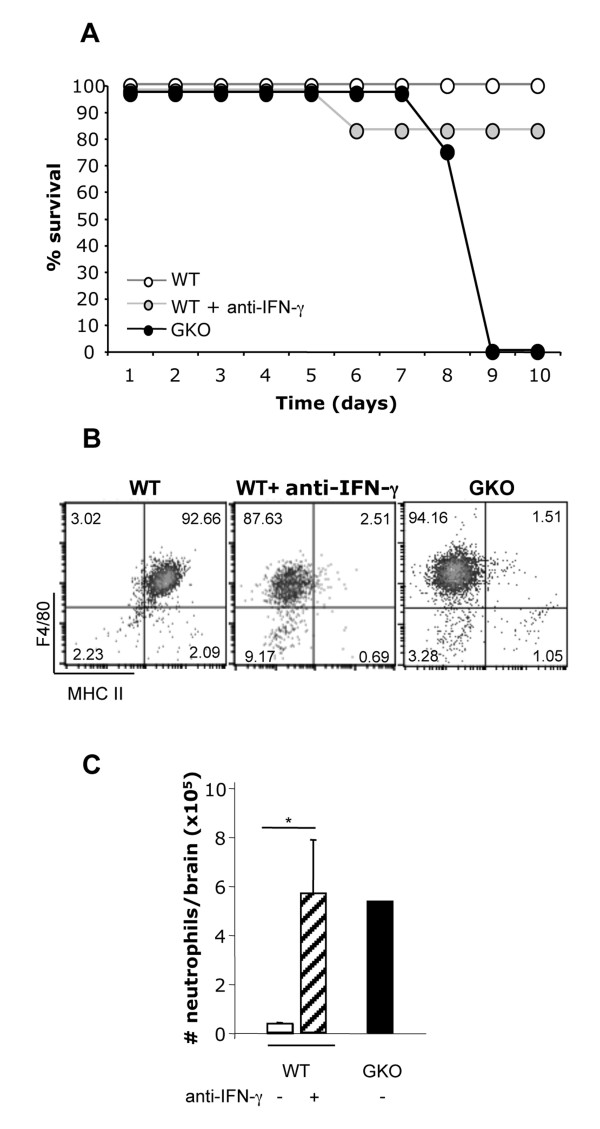
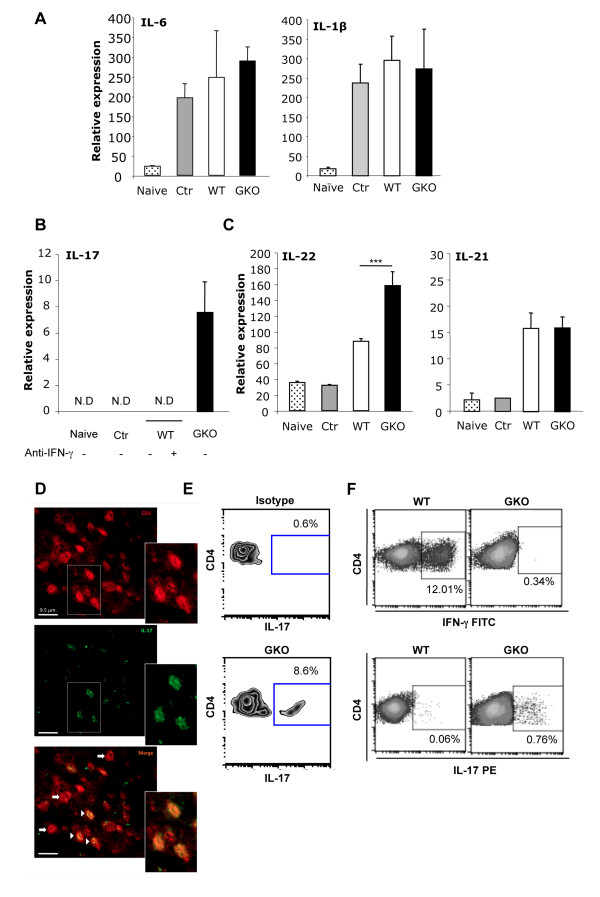
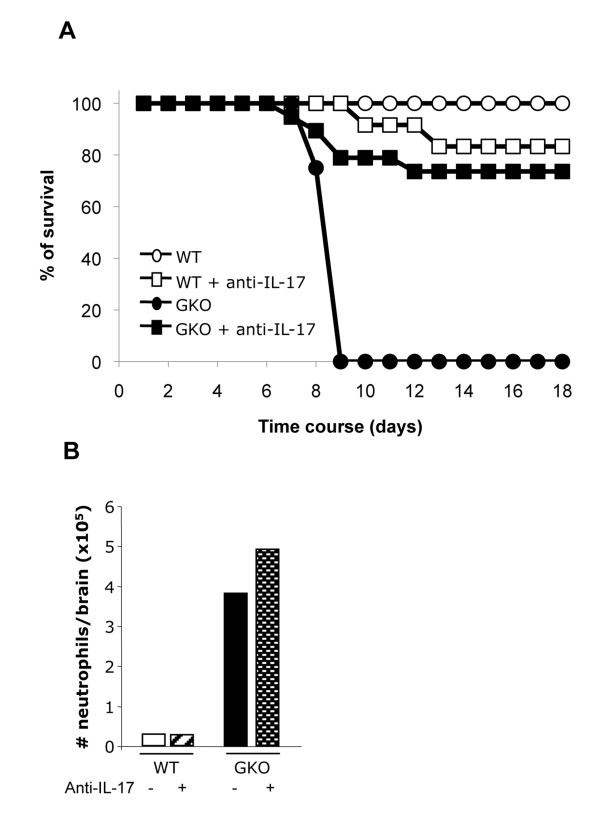
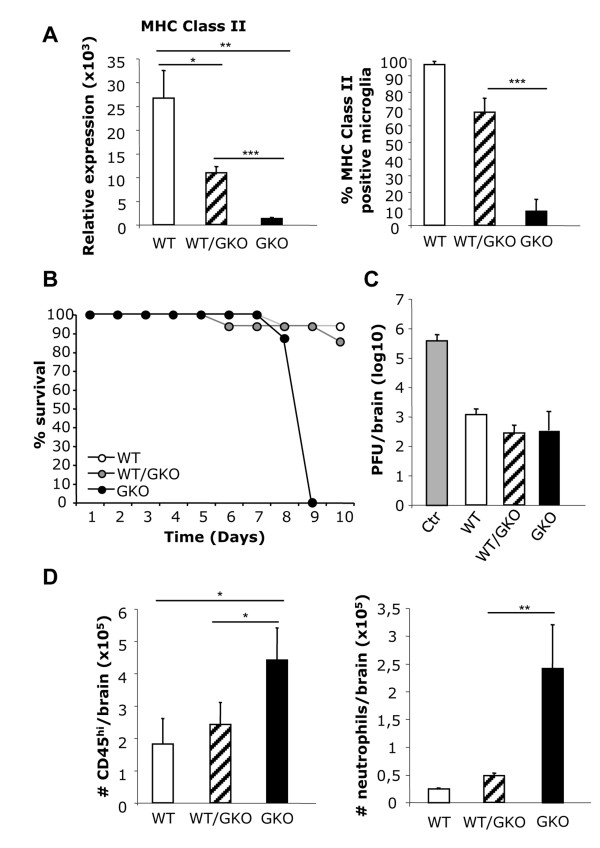
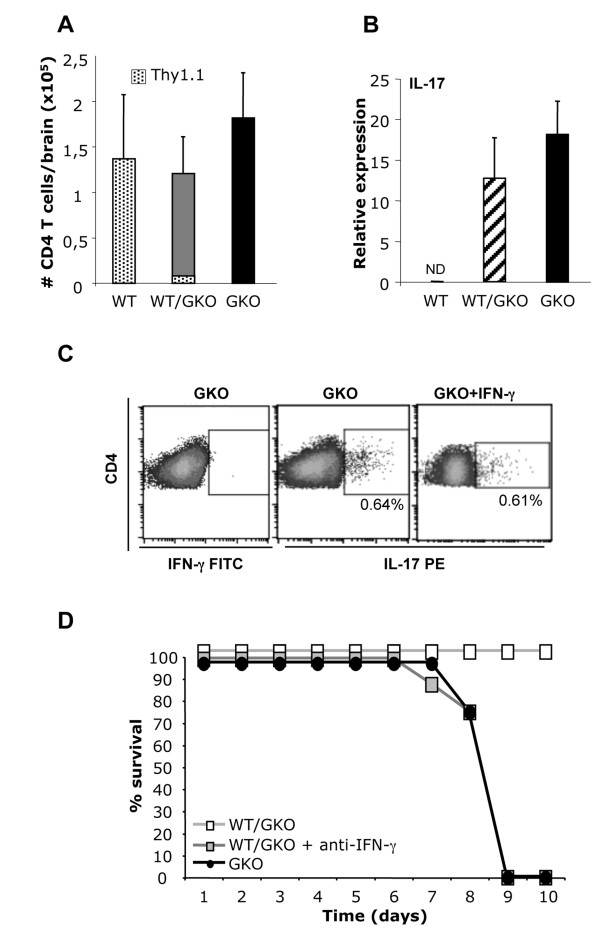
Transfer of GKO memory CD4+ T cells into infected SCID mice induced rapid mortality compared to recipients of WT memory CD4+ T cells, despite similar virus control and demyelination. In contrast to recipients of WT CD4+ T cells, extensive neutrophil infiltration and IL-17 expression within the CNS in recipients of GKO CD4+ T cells provided a model to directly assess their contribution(s) to disease. Recipients of WT CD4+ T cells depleted of IFN-γ did not express IL-17 and were spared from mortality despite abundant CNS neutrophil infiltration, indicating that mortality was not mediated by excessive CNS neutrophil accumulation. By contrast, IL-17 depletion rescued recipients of GKO CD4+ T cells from rapid mortality without diminishing neutrophils or reducing GM-CSF, associated with pathogenic Th17 cells in CNS autoimmune models. Furthermore, co-transfer of WT and GKO CD4+ T cells prolonged survival in an IFN-γ dependent manner, although IL-17 transcription was not reduced.
These data demonstrate that IL-17 mediates detrimental clinical consequences in an IFN-γ-deprived environment, independent of extensive neutrophil accumulation or GM-CSF upregulation. The results also suggest that IFN-γ overrides the detrimental IL-17 effector responses via a mechanism downstream of transcriptional regulation.
IFN-γ、IL-17 和中性粒细胞在中枢神经系统炎症性疾病中的相互作用非常复杂,因为交叉调节因子会影响正反馈和负反馈回路。这些相互作用阻碍了区分中性粒细胞、Th1 和 Th17 细胞衍生的效应分子以及次级介质对组织损伤和发病率的相对贡献的能力。
使用嗜神经鼠冠状病毒诱导的脑炎作为模型,评估中性粒细胞、IFN-γ 和 IL-17 对病毒诱导的死亡率的直接贡献。通过将病毒预先激活的野生型(WT)或 IFN-γ 缺陷(GKO)记忆 CD4+T 细胞过继转移到感染的 SCID 小鼠中,同时进行抗体介导的中性粒细胞耗竭和细胞因子阻断,来选择性地操纵中枢神经系统炎症状态。
与接受 WT 记忆 CD4+T 细胞的受体相比,将 GKO 记忆 CD4+T 细胞转移到感染的 SCID 小鼠中会导致快速死亡,尽管病毒控制和脱髓鞘相似。与接受 WT CD4+T 细胞的受体不同,在接受 GKO CD4+T 细胞的受体中,中枢神经系统内广泛的中性粒细胞浸润和 IL-17 表达提供了一个直接评估其对疾病贡献的模型。尽管中枢神经系统中性粒细胞浸润丰富,但接受 WT CD4+T 细胞耗竭 IFN-γ 的受体不表达 IL-17,且免于死亡,表明死亡率不是由中枢神经系统中性粒细胞过度积累介导的。相比之下,IL-17 耗竭使接受 GKO CD4+T 细胞的受体免于快速死亡,而不会减少中性粒细胞或降低 GM-CSF,与中枢神经系统自身免疫模型中的致病性 Th17 细胞相关。此外,WT 和 GKO CD4+T 细胞的共转移以 IFN-γ 依赖的方式延长了存活时间,尽管 IL-17 转录没有减少。
这些数据表明,IL-17 在 IFN-γ 缺乏的环境中介导有害的临床后果,与广泛的中性粒细胞积累或 GM-CSF 上调无关。结果还表明,IFN-γ 通过一种转录后调节的机制,抑制有害的 IL-17 效应反应。