Department of Molecular Immunology, Beckman Research Institute of City of Hope, Duarte, California, United States of America.
PLoS Pathog. 2018 Jan 19;14(1):e1006822. doi: 10.1371/journal.ppat.1006822. eCollection 2018 Jan.
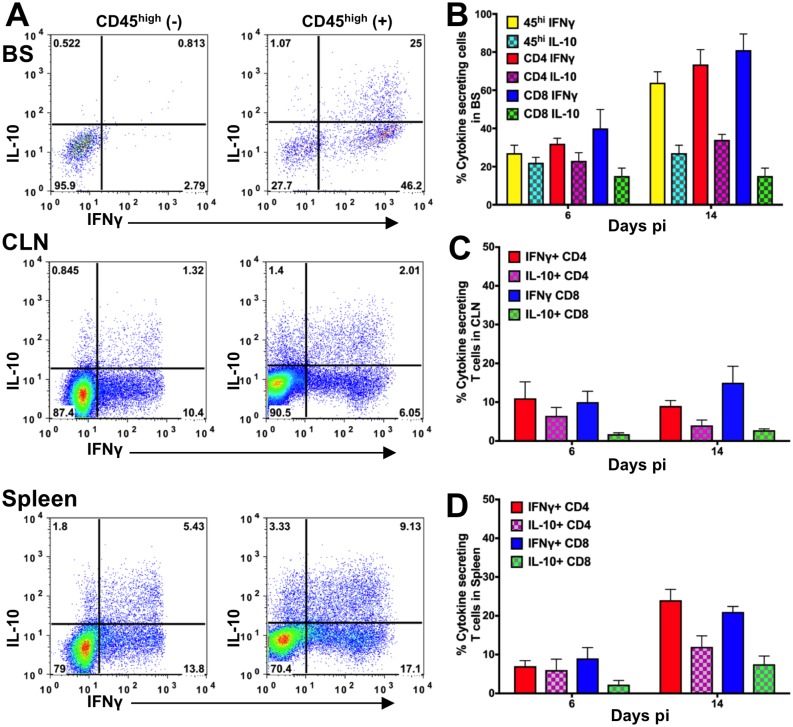
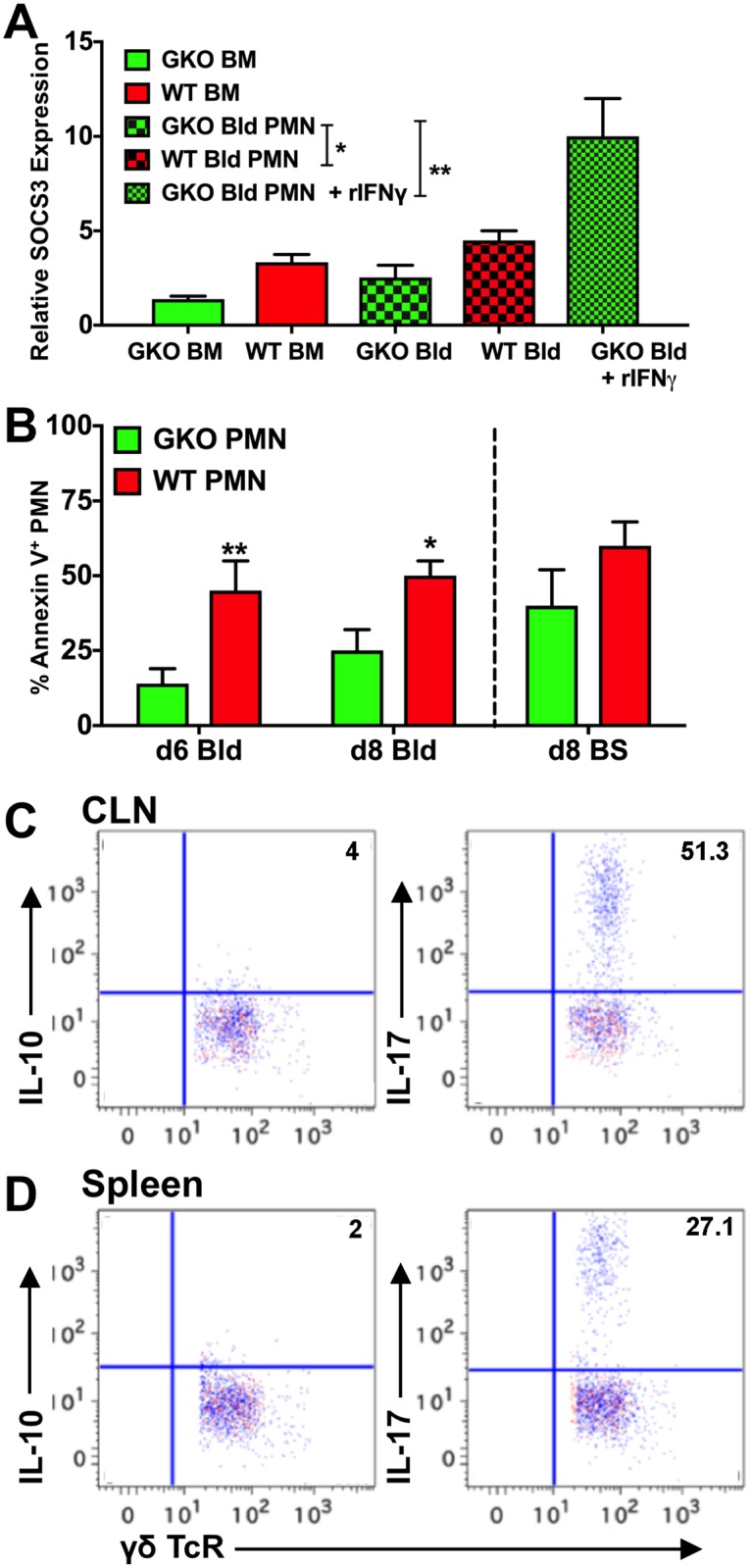
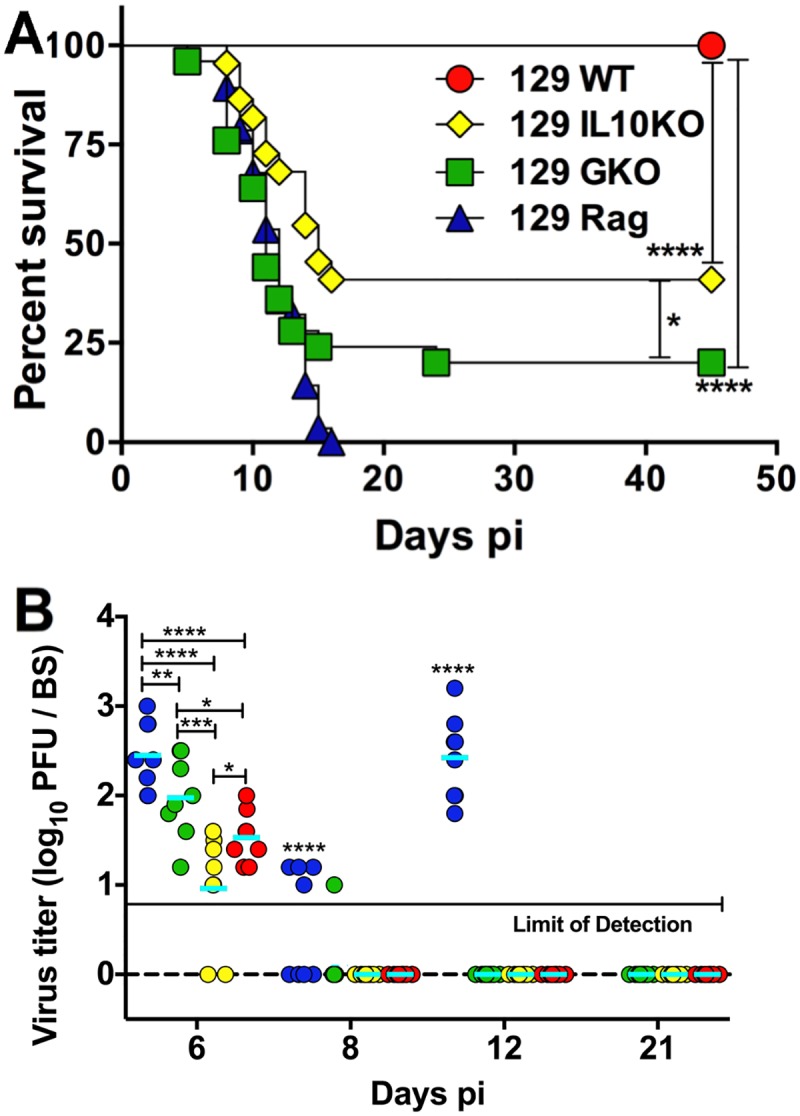
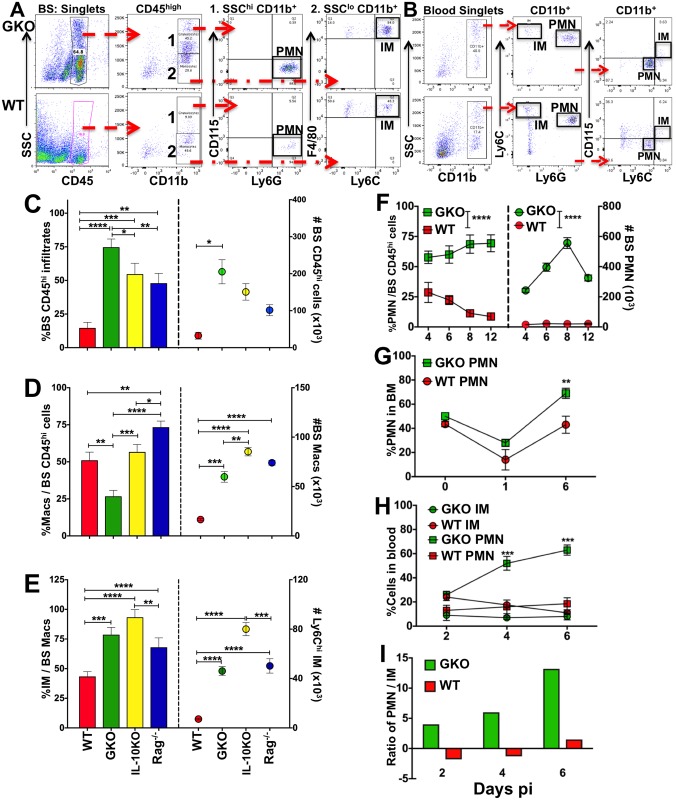
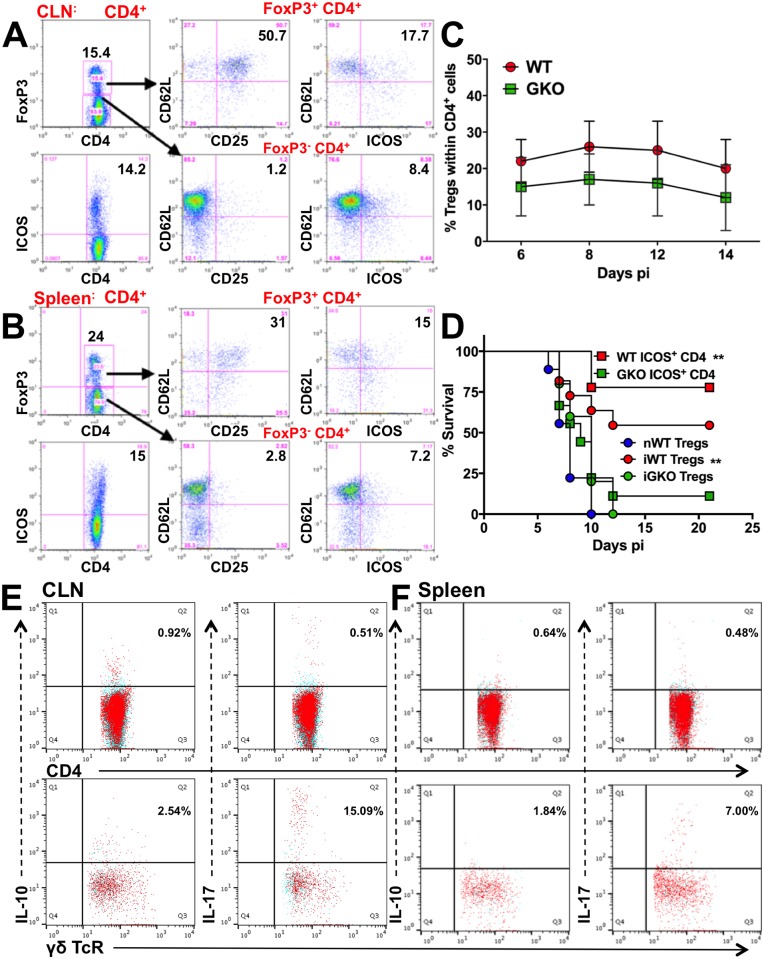
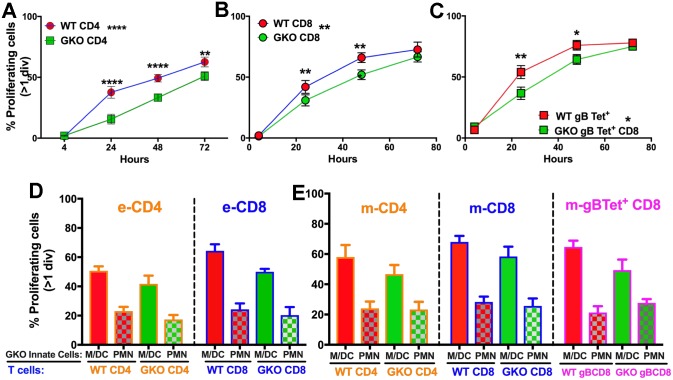
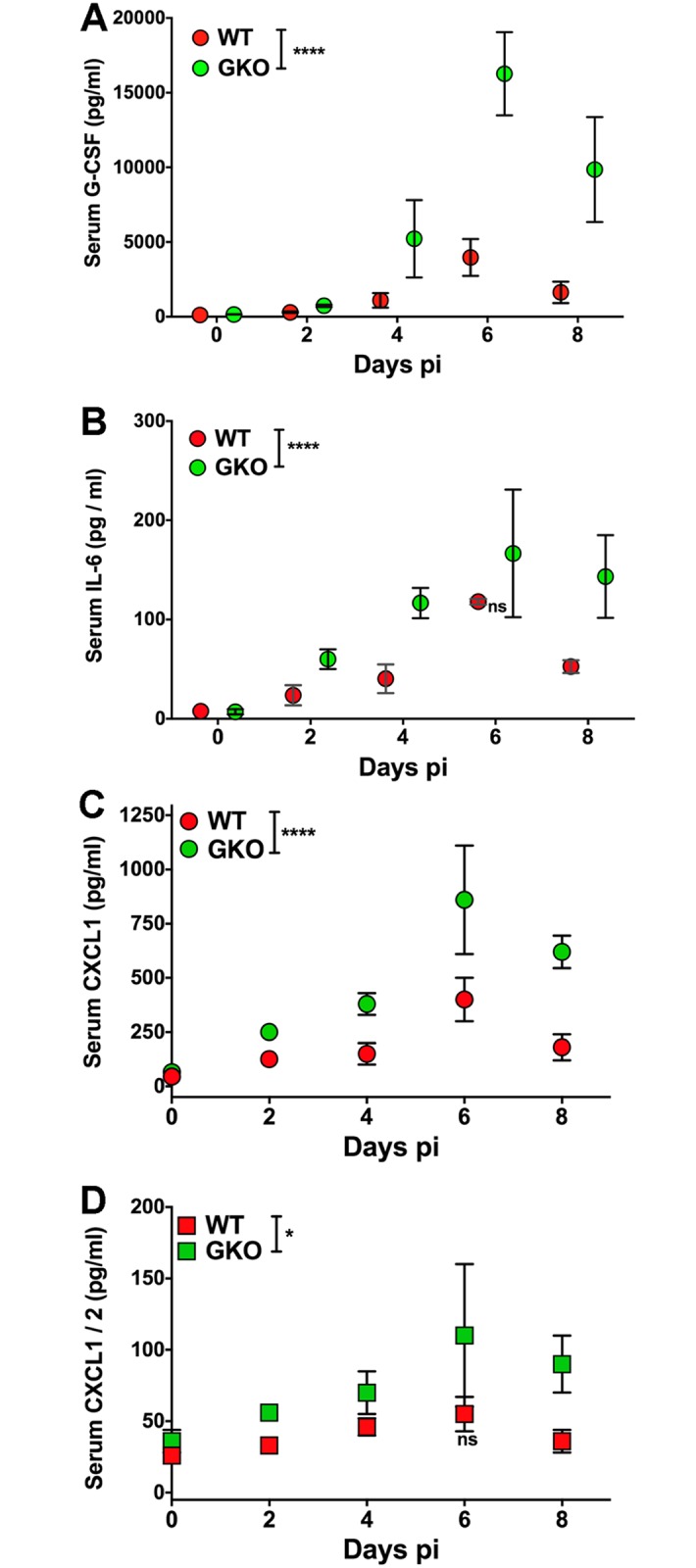
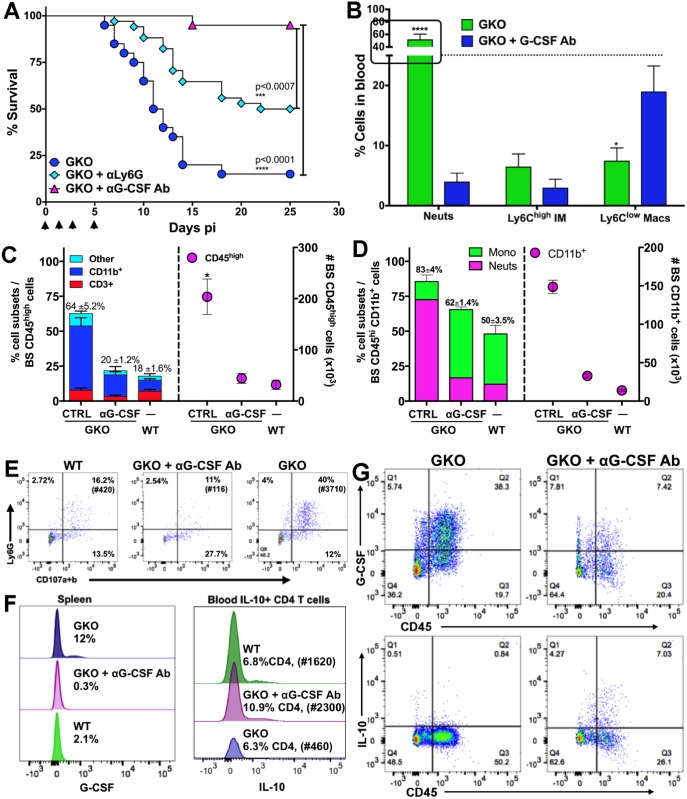
Emergency hematopoiesis facilitates the rapid expansion of inflammatory immune cells in response to infections by pathogens, a process that must be carefully regulated to prevent potentially life threatening inflammatory responses. Here, we describe a novel regulatory role for the cytokine IFNγ that is critical for preventing fatal encephalitis after viral infection. HSV1 encephalitis (HSE) is triggered by the invasion of the brainstem by inflammatory monocytes and neutrophils. In mice lacking IFNγ (GKO), we observed unrestrained increases in G-CSF levels but not in GM-CSF or IL-17. This resulted in uncontrolled expansion and infiltration of apoptosis-resistant, degranulating neutrophils into the brainstem, causing fatal HSE in GKO but not WT mice. Excessive G-CSF in GKO mice also induced granulocyte derived suppressor cells, which inhibited T-cell proliferation and function, including production of the anti-inflammatory cytokine IL-10. Unexpectedly, we found that IFNγ suppressed G-CSF signaling by increasing SOCS3 expression in neutrophils, resulting in apoptosis. Depletion of G-CSF, but not GM-CSF, in GKO mice induced neutrophil apoptosis and reinstated IL-10 secretion by T cells, which restored their ability to limit innate inflammatory responses resulting in protection from HSE. Our studies reveals a novel, complex interplay among IFNγ, G-CSF and IL-10, which highlights the opposing roles of G-CSF and IFNγ in regulation of innate inflammatory responses in a murine viral encephalitis model and reveals G-CSF as a potential therapeutic target. Thus, the antagonistic G-CSF-IFNγ interactions emerge as a key regulatory node in control of CNS inflammatory responses to virus infection.
应急造血促进了炎症免疫细胞在应对病原体感染时的快速扩张,这一过程必须得到精心调节,以防止潜在的危及生命的炎症反应。在这里,我们描述了细胞因子 IFNγ 的一个新的调节作用,它对于防止病毒感染后的致命性脑炎是至关重要的。HSV1 脑炎(HSE)是由炎症性单核细胞和中性粒细胞侵袭脑干引发的。在缺乏 IFNγ(GKO)的小鼠中,我们观察到 G-CSF 水平不受控制地增加,但 GM-CSF 或 IL-17 没有增加。这导致凋亡抵抗、脱颗粒的中性粒细胞不受控制地扩张和浸润到脑干中,导致 GKO 小鼠而不是 WT 小鼠发生致命性 HSE。GKO 小鼠中过多的 G-CSF 还诱导了粒细胞衍生的抑制细胞,抑制了 T 细胞的增殖和功能,包括抗炎细胞因子 IL-10 的产生。出乎意料的是,我们发现 IFNγ 通过增加中性粒细胞中 SOCS3 的表达来抑制 G-CSF 信号,导致细胞凋亡。在 GKO 小鼠中耗尽 G-CSF,但不是 GM-CSF,诱导中性粒细胞凋亡,并恢复 T 细胞分泌 IL-10,恢复其限制固有炎症反应的能力,从而防止 HSE。我们的研究揭示了 IFNγ、G-CSF 和 IL-10 之间的一种新的、复杂的相互作用,突出了 G-CSF 和 IFNγ 在调节小鼠病毒性脑炎模型中固有炎症反应中的相反作用,并揭示了 G-CSF 作为一个潜在的治疗靶点。因此,G-CSF-IFNγ 的相互作用作为控制中枢神经系统对病毒感染的炎症反应的关键调节节点出现。