Bioinformatics Graduate Program, University of California, San Diego, La Jolla, CA 92093, USA.
Bioinformatics. 2012 Jun 15;28(12):i188-96. doi: 10.1093/bioinformatics/bts219.
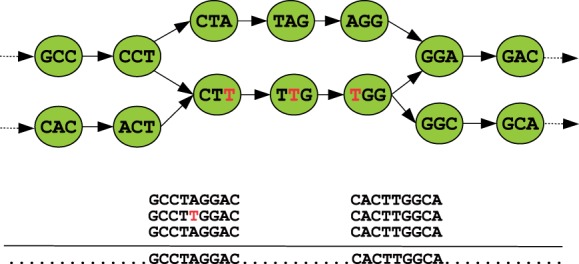
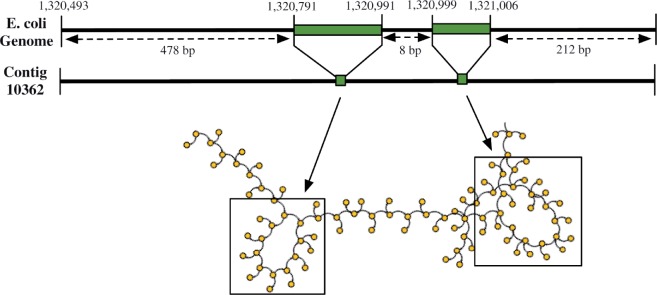
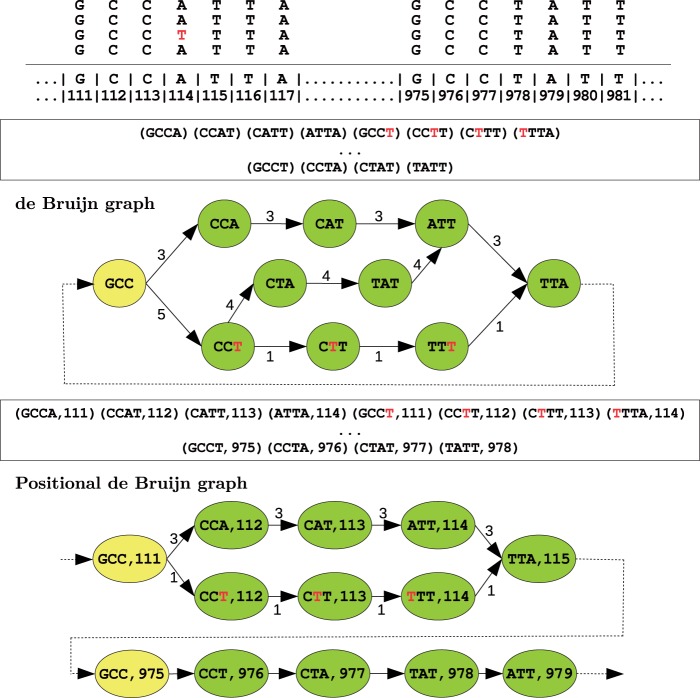
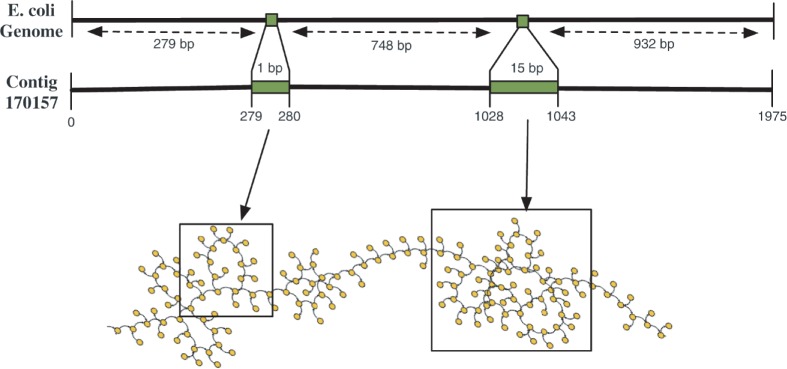
Assemblies of next-generation sequencing (NGS) data, although accurate, still contain a substantial number of errors that need to be corrected after the assembly process. We develop SEQuel, a tool that corrects errors (i.e. insertions, deletions and substitution errors) in the assembled contigs. Fundamental to the algorithm behind SEQuel is the positional de Bruijn graph, a graph structure that models k-mers within reads while incorporating the approximate positions of reads into the model.
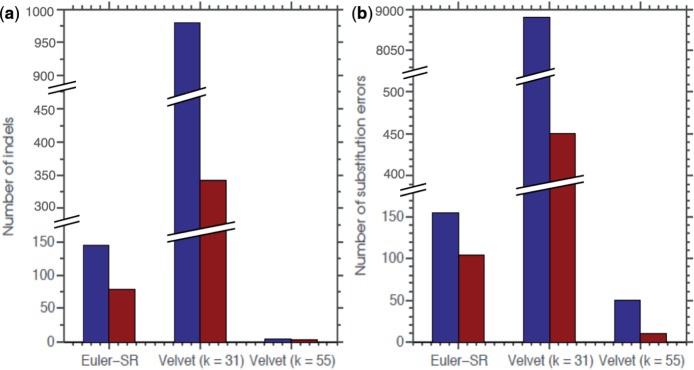
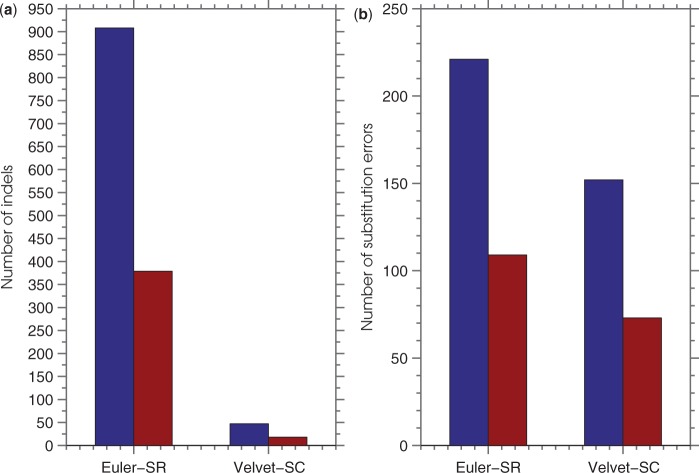
SEQuel reduced the number of small insertions and deletions in the assemblies of standard multi-cell Escherichia coli data by almost half, and corrected between 30% and 94% of the substitution errors. Further, we show SEQuel is imperative to improving single-cell assembly, which is inherently more challenging due to higher error rates and non-uniform coverage; over half of the small indels, and substitution errors in the single-cell assemblies were corrected. We apply SEQuel to the recently assembled Deltaproteobacterium SAR324 genome, which is the first bacterial genome with a comprehensive single-cell genome assembly, and make over 800 changes (insertions, deletions and substitutions) to refine this assembly.
SEQuel can be used as a post-processing step in combination with any NGS assembler and is freely available at http://bix.ucsd.edu/SEQuel/.
尽管下一代测序 (NGS) 数据的组装结果很准确,但仍包含大量需要在组装过程后进行纠正的错误。我们开发了 SEQuel,这是一种可纠正组装后重叠群中错误(即插入、缺失和替换错误)的工具。SEQuel 背后的算法的基础是位置 de Bruijn 图,这是一种在读取内容中对 k-mers 进行建模的图结构,同时将读取的近似位置纳入模型中。
SEQuel 将标准多细胞大肠杆菌数据的组装中较小的插入和缺失数量减少了近一半,并纠正了 30%至 94%的替换错误。此外,我们还表明,SEQuel 对于改进单细胞组装至关重要,因为其错误率更高且覆盖不均匀,因此本身更具挑战性;单细胞组装中的一半以上的小插入和替换错误都得到了纠正。我们将 SEQuel 应用于最近组装的δ变形菌 SAR324 基因组,这是第一个具有全面单细胞基因组组装的细菌基因组,并对其进行了 800 多次修改(插入、缺失和替换)以完善该组装。
SEQuel 可以作为任何 NGS 组装器的后处理步骤使用,可在 http://bix.ucsd.edu/SEQuel/ 免费获取。