Centro de Biología Molecular Severo Ochoa (CBMSO) (CSIC-UAM), Madrid, Spain.
Brief Bioinform. 2021 Nov 5;22(6). doi: 10.1093/bib/bbab170.
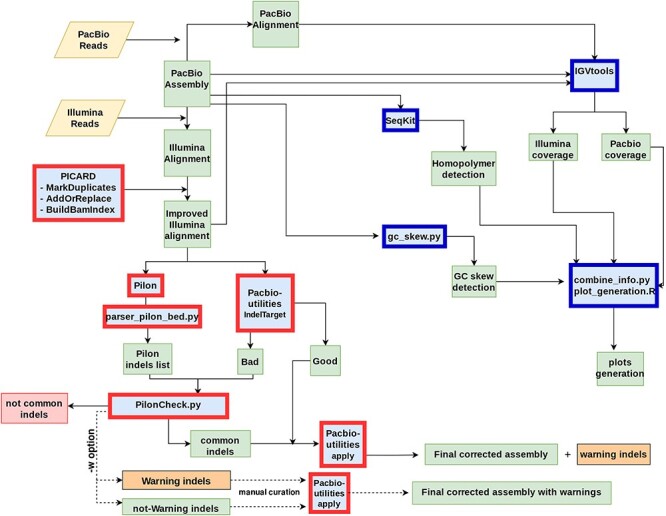
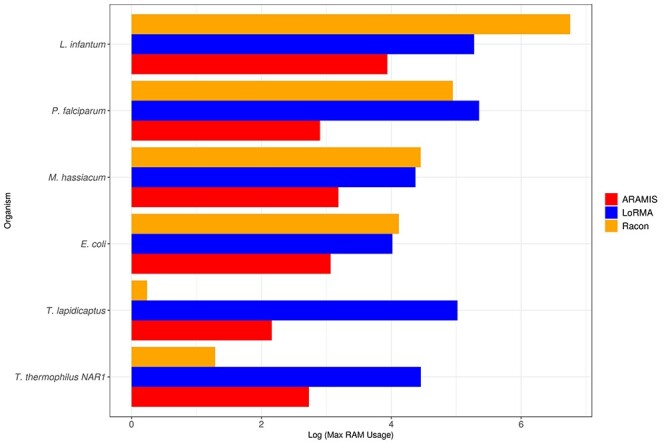
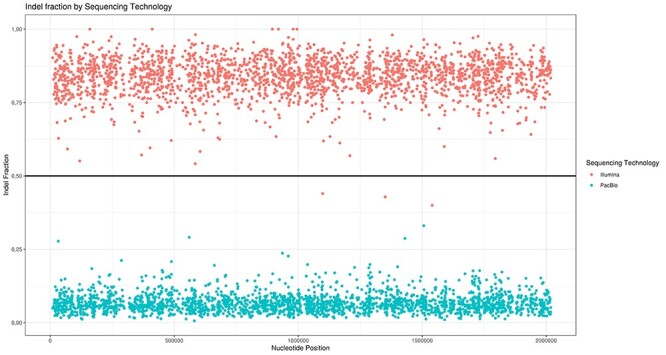
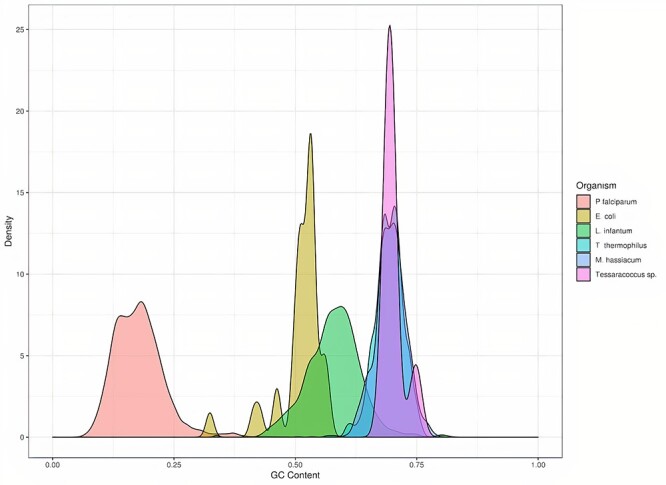
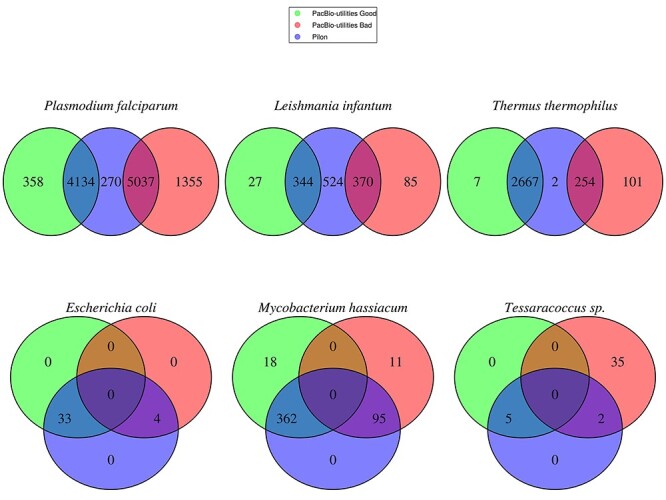
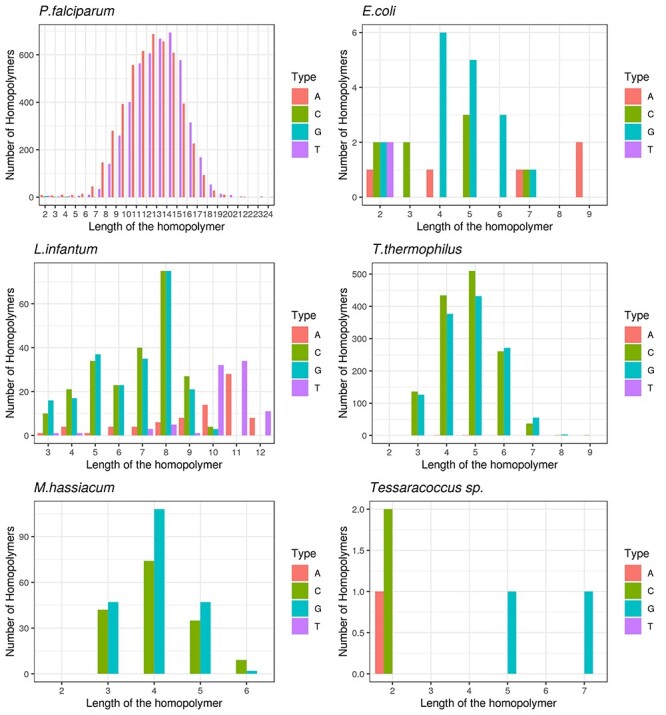
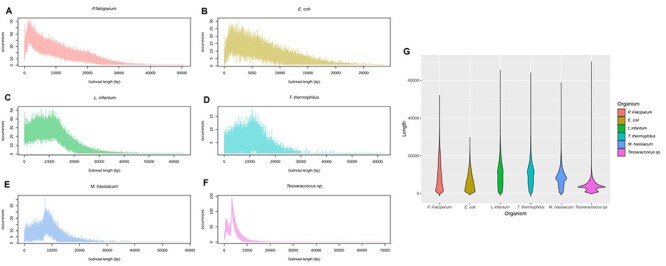
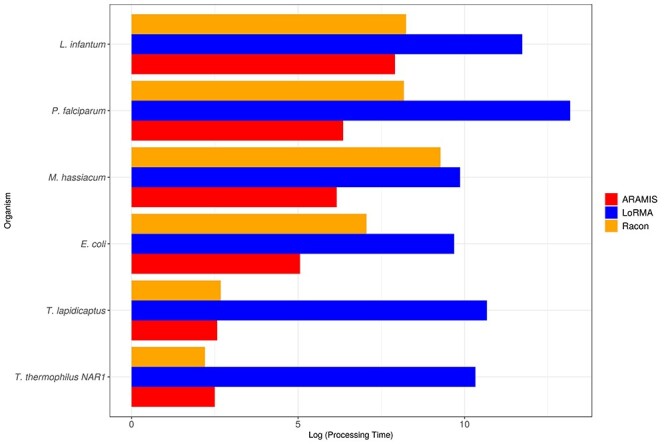
NGS long-reads sequencing technologies (or third generation) such as Pacific BioSciences (PacBio) have revolutionized the sequencing field over the last decade improving multiple genomic applications like de novo genome assemblies. However, their error rate, mostly involving insertions and deletions (indels), is currently an important concern that requires special attention to be solved. Multiple algorithms are available to fix these sequencing errors using short reads (such as Illumina), although they require long processing times and some errors may persist. Here, we present Accurate long-Reads Assembly correction Method for Indel errorS (ARAMIS), the first NGS long-reads indels correction pipeline that combines several correction software in just one step using accurate short reads. As a proof OF concept, six organisms were selected based on their different GC content, size and genome complexity, and their PacBio-assembled genomes were corrected thoroughly by this pipeline. We found that the presence of systematic sequencing errors in long-reads PacBio sequences affecting homopolymeric regions, and that the type of indel error introduced during PacBio sequencing are related to the GC content of the organism. The lack of knowledge of this fact leads to the existence of numerous published studies where such errors have been found and should be resolved since they may contain incorrect biological information. ARAMIS yields better results with less computational resources needed than other correction tools and gives the possibility of detecting the nature of the found indel errors found and its distribution along the genome. The source code of ARAMIS is available at https://github.com/genomics-ngsCBMSO/ARAMIS.git.
NGS 长读测序技术(或第三代),如 PacificBioSciences(PacBio),在过去十年中彻底改变了测序领域,改善了从头基因组组装等多种基因组应用。然而,它们的错误率,主要涉及插入和缺失(indels),目前是一个需要特别关注的重要问题。有多种算法可用于使用短读(如 Illumina)来修复这些测序错误,尽管它们需要较长的处理时间,并且一些错误可能仍然存在。在这里,我们提出了用于 indel 错误的准确长读组装校正方法(ARAMIS),这是第一个 NGS 长读 indels 校正管道,它使用准确的短读在一步中结合了几种校正软件。作为概念验证,我们根据不同的 GC 含量、大小和基因组复杂性选择了六个生物体,并通过该管道彻底校正了它们的 PacBio 组装基因组。我们发现,长读 PacBio 序列中存在系统的测序错误,影响了同源多聚区域,并且 PacBio 测序过程中引入的 indel 错误类型与生物体的 GC 含量有关。由于缺乏对这一事实的了解,导致了许多发表的研究中都发现了此类错误,并且应该加以解决,因为它们可能包含不正确的生物学信息。与其他校正工具相比,ARAMIS 所需的计算资源更少,但结果更好,并提供了检测发现的 indel 错误的性质及其在基因组中的分布的可能性。ARAMIS 的源代码可在 https://github.com/genomics-ngsCBMSO/ARAMIS.git 获得。