Center for Theoretical Biology, Peking University, Beijing 100871, People's Republic of China.
BMC Med Genomics. 2012 Jun 12;5:24. doi: 10.1186/1755-8794-5-24.
Copy number variation (CNV) is essential to understand the pathology of many complex diseases at the DNA level. Affymetrix SNP arrays, which are widely used for CNV studies, significantly depend on accurate copy number (CN) estimation. Nevertheless, CN estimation may be biased by several factors, including cross-hybridization and training sample batch, as well as genomic waves of intensities induced by sequence-dependent hybridization rate and amplification efficiency. Since many available algorithms only address one or two of the three factors, a high false discovery rate (FDR) often results when identifying CNV. Therefore, we have developed a new CNV detection pipeline which is based on hybridization and amplification rate correction (CNVhac).
CNVhac first estimates the allelic concentrations (ACs) of target sequences by using the sample independent parameters trained through physicochemical hybridization law. Then the raw CN is estimated by taking the ratio of AC to the corresponding average AC from a reference sample set for one specific site. Finally, a hidden Markov model (HMM) segmentation process is implemented to detect CNV regions.
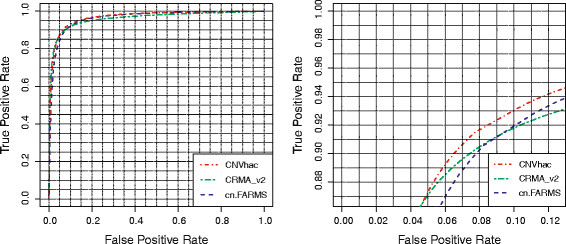
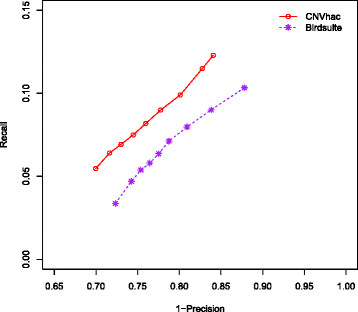
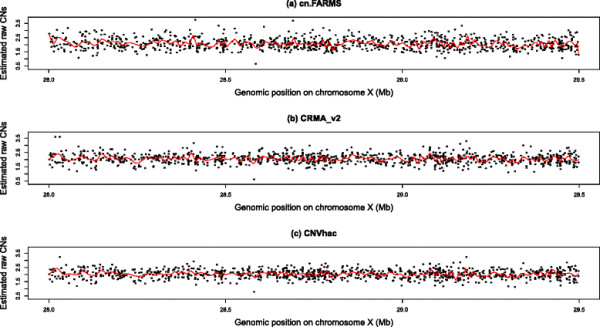
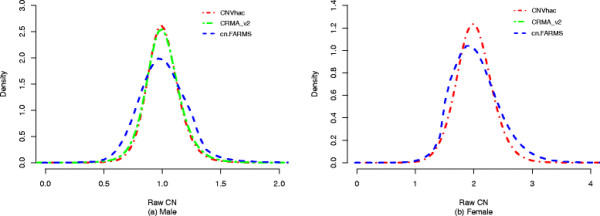
Based on public HapMap data, the results show that CNVhac effectively smoothes the genomic waves and facilitates more accurate raw CN estimates compared to other methods. Moreover, CNVhac alleviates, to a certain extent, the sample dependence of inference and makes CNV calling with appreciable low FDRs.
CNVhac is an effective approach to address the common difficulties in SNP array analysis, and the working principles of CNVhac can be easily extended to other platforms.
拷贝数变异(CNV)对于理解许多复杂疾病的 DNA 水平的病理学至关重要。Affymetrix SNP 阵列广泛用于 CNV 研究,其对准确的拷贝数(CN)估计有很大的依赖性。然而,CN 估计可能会受到多种因素的影响,包括交叉杂交和训练样本批次,以及序列依赖性杂交率和扩增效率引起的基因组强度波动。由于许多可用的算法仅解决三个因素中的一个或两个因素,因此在识别 CNV 时通常会导致高假发现率(FDR)。因此,我们开发了一种新的 CNV 检测管道,该管道基于杂交和扩增率校正(CNVhac)。
CNVhac 首先通过使用通过物理化学杂交律训练的样本独立参数来估计目标序列的等位基因浓度(AC)。然后,通过将 AC 与参考样本集中特定位置的相应平均 AC 的比值来估计原始 CN。最后,实施隐马尔可夫模型(HMM)分割过程来检测 CNV 区域。
基于公共 HapMap 数据,结果表明,与其他方法相比,CNVhac 有效地平滑了基因组波动,并且更准确地估计了原始 CN。此外,CNVhac 在一定程度上减轻了推断的样本依赖性,并使 CNV 调用具有可察觉的低 FDR。
CNVhac 是解决 SNP 阵列分析中常见困难的有效方法,并且 CNVhac 的工作原理可以很容易地扩展到其他平台。