Department of Chemistry and Institute for Computational Molecular Science, Temple University, Philadelphia, Pennsylvania, United States of America.
PLoS Comput Biol. 2012 May;8(5):e1002532. doi: 10.1371/journal.pcbi.1002532. Epub 2012 May 31.
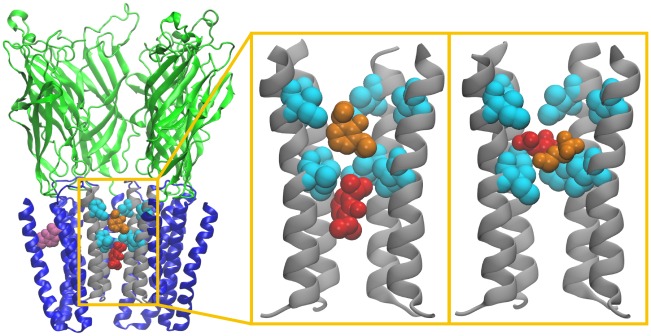
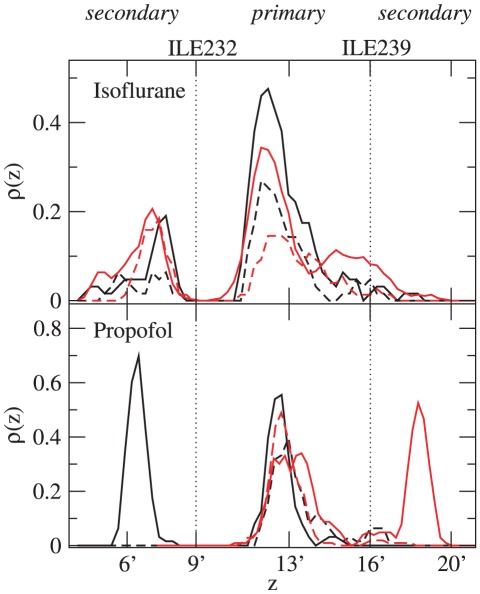
Although general anesthetics are known to modulate the activity of ligand-gated ion channels in the Cys-loop superfamily, there is at present neither consensus on the underlying mechanisms, nor predictive models of this modulation. Viable models need to offer quantitative assessment of the relative importance of several identified anesthetic binding sites. However, to date, precise affinity data for individual sites has been challenging to obtain by biophysical means. Here, the likely role of pore block inhibition by the general anesthetics isoflurane and propofol of the prokaryotic pentameric channel GLIC is investigated by molecular simulations. Microscopic affinities are calculated for both single and double occupancy binding of isoflurane and propofol to the GLIC pore. Computations are carried out for an open-pore conformation in which the pore is restrained to crystallographic radius, and a closed-pore conformation that results from unrestrained molecular dynamics equilibration of the structure. The GLIC pore is predicted to be blocked at the micromolar concentrations for which inhibition by isofluorane and propofol is observed experimentally. Calculated affinities suggest that pore block by propofol occurs at signifcantly lower concentrations than those for which inhibition is observed: we argue that this discrepancy may result from binding of propofol to an allosteric site recently identified by X-ray crystallography, which may cause a competing gain-of-function effect. Affinities of isoflurane and propofol to the allosteric site are also calculated, and shown to be 3 mM for isoflurane and 10 μM for propofol; both anesthetics have a lower affinity for the allosteric site than for the unoccupied pore.
虽然全身麻醉剂已知可调节 Cys 环超家族中配体门控离子通道的活性,但目前对于这种调节的潜在机制尚未达成共识,也没有预测模型。可行的模型需要对几个已识别的麻醉剂结合位点的相对重要性进行定量评估。然而,迄今为止,通过生物物理手段获得单个位点的精确亲和力数据一直具有挑战性。在这里,通过分子模拟研究了全身麻醉剂异氟烷和丙泊酚对原核五聚体通道 GLIC 的孔阻塞抑制的可能作用。计算了异氟烷和丙泊酚单占据和双占据 GLIC 孔的微观亲和力。计算是针对开放孔构象进行的,其中孔被限制在晶体半径,以及由结构的无约束分子动力学平衡产生的闭合孔构象。GLIC 孔预计在微摩尔浓度下被阻塞,在该浓度下观察到异氟烷和丙泊酚的抑制作用。计算出的亲和力表明,丙泊酚的孔阻塞发生在明显低于观察到抑制作用的浓度:我们认为这种差异可能是由于丙泊酚结合最近通过 X 射线晶体学鉴定的变构位点引起的,这可能导致竞争的功能获得效应。还计算了异氟烷和丙泊酚与变构位点的亲和力,结果表明异氟烷的亲和力为 3 mM,丙泊酚的亲和力为 10 μM;两种麻醉剂与未占据的孔相比,对变构位点的亲和力都较低。