Robert S. Boas Center for Human Genetics and Genomics, The Feinstein Institute for Medical Research, Northshore LIJ Healthsystem, Manhasset, New York, United States of America.
PLoS One. 2012;7(6):e38087. doi: 10.1371/journal.pone.0038087. Epub 2012 Jun 6.
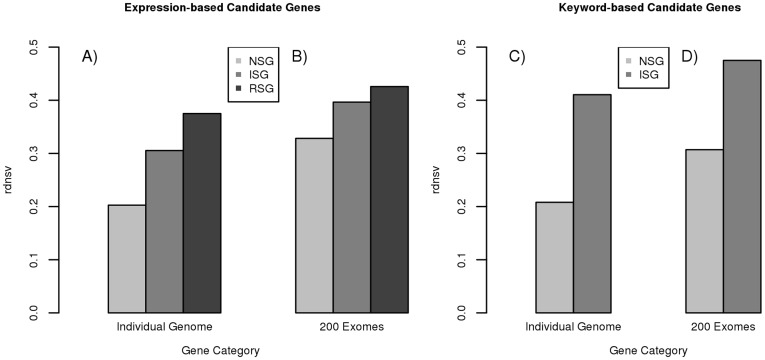
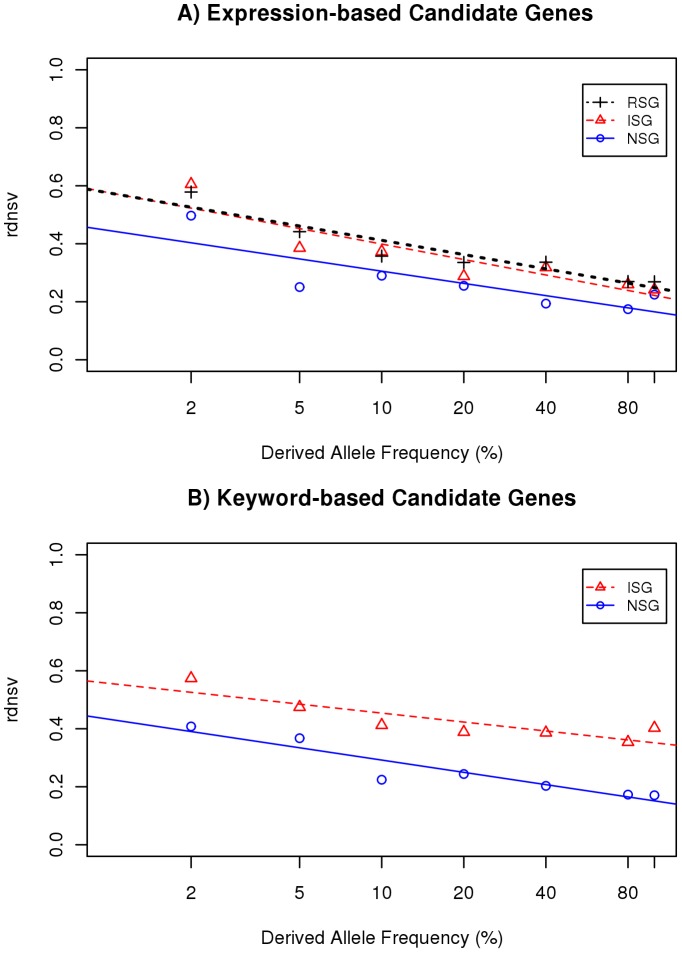
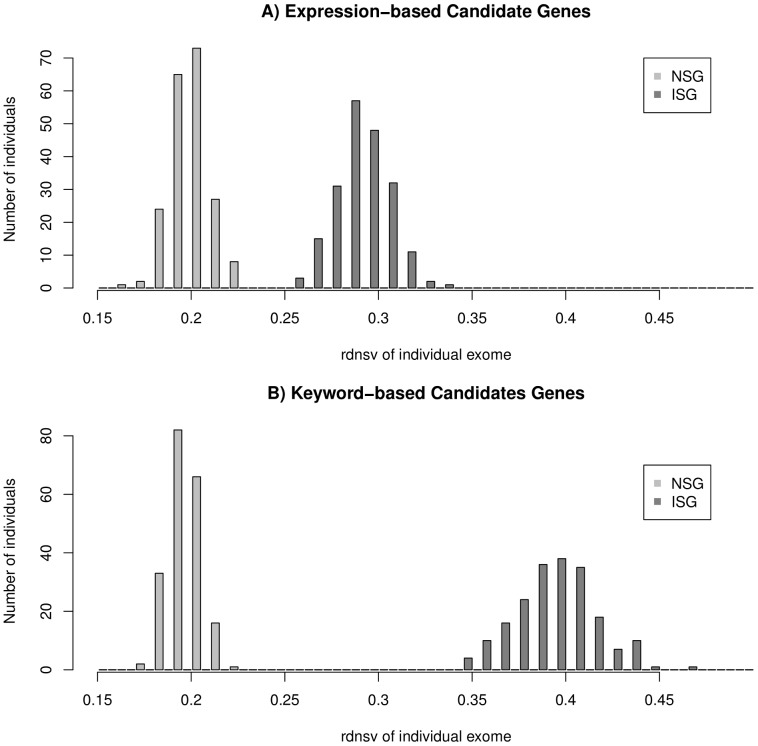
To measure the strength of natural selection that acts upon single nucleotide variants (SNVs) in a set of human genes, we calculate the ratio between nonsynonymous SNVs (nsSNVs) per nonsynonymous site and synonymous SNVs (sSNVs) per synonymous site. We transform this ratio with a respective factor f that corrects for the bias of synonymous sites towards transitions in the genetic code and different mutation rates for transitions and transversions. This method approximates the relative density of nsSNVs (rdnsv) in comparison with the neutral expectation as inferred from the density of sSNVs. Using SNVs from a diploid genome and 200 exomes, we apply our method to immune system genes (ISGs), nervous system genes (NSGs), randomly sampled genes (RSGs), and gene ontology annotated genes. The estimate of rdnsv in an individual exome is around 20% for NSGs and 30-40% for ISGs and RSGs. This smaller rdnsv of NSGs indicates overall stronger purifying selection. To quantify the relative shift of nsSNVs towards rare variants, we next fit a linear regression model to the estimates of rdnsv over different SNV allele frequency bins. The obtained regression models show a negative slope for NSGs, ISGs and RSGs, supporting an influence of purifying selection on the frequency spectrum of segregating nsSNVs. The y-intercept of the model predicts rdnsv for an allele frequency close to 0. This parameter can be interpreted as the proportion of nonsynonymous sites where mutations are tolerated to segregate with an allele frequency notably greater than 0 in the population, given the performed normalization of the observed nsSNV to sSNV ratio. A smaller y-intercept is displayed by NSGs, indicating more nonsynonymous sites under strong negative selection. This predicts more monogenically inherited or de-novo mutation diseases that affect the nervous system.
为了衡量在一组人类基因中单核苷酸变异(SNV)所受自然选择的强度,我们计算非同义 SNV(nsSNV)与非同义位点之比以及同义 SNV(sSNV)与同义位点之比。我们用一个相应的因子 f 对该比值进行变换,该因子 f 校正遗传密码中同义位点向转换的偏向性以及转换和颠换的不同突变率。这种方法近似于与同义 SNV 密度相比 nsSNV 的相对密度(rdnsv),中性预期是根据同义 SNV 的密度推断出来的。我们使用来自二倍体基因组和 200 个外显子的 SNV,将我们的方法应用于免疫系统基因(ISGs)、神经系统基因(NSGs)、随机抽样基因(RSGs)和基因本体注释基因。个体外显子中 rdnsv 的估计值约为 NSGs 的 20%和 ISGs 和 RSGs 的 30-40%。NSGs 中 rdnsv 较小表明整体净化选择更强。为了量化 nsSNV 向稀有变异的相对转移,我们接下来对 rdnsv 在不同 SNV 等位基因频率箱中的估计值拟合线性回归模型。获得的回归模型显示 NSGs、ISGs 和 RSGs 的斜率为负,支持净化选择对分离 nsSNV 频率谱的影响。模型的 y 截距预测了等位基因频率接近 0 时的 rdnsv。该参数可以解释为在进行观测到的 nsSNV 与 sSNV 比值的归一化后,在群体中具有显著大于 0 的等位基因频率分离的情况下,容忍突变的非同义位点的比例。NSGs 的 y 截距较小,表明有更多的非同义位点受到强烈的负选择。这预测了更多影响神经系统的单基因遗传或新生突变疾病。