Palo Alto Research Center, 3333 Coyote Hill Road, Palo Alto, California 94304, United States.
J Proteome Res. 2012 Aug 3;11(8):4191-200. doi: 10.1021/pr300312h. Epub 2012 Jul 2.
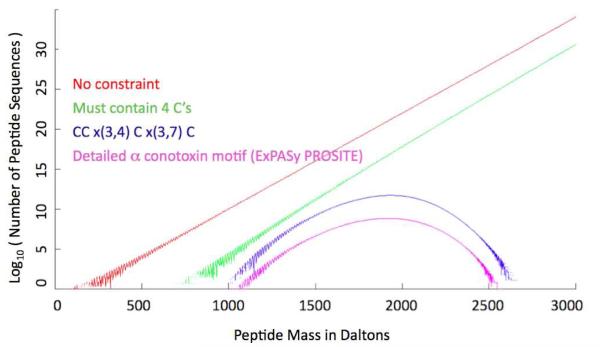
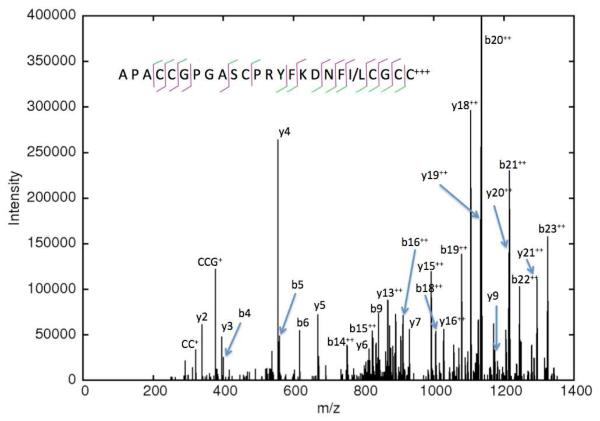
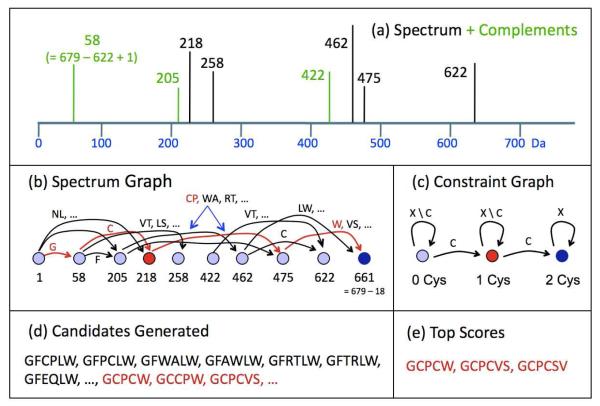
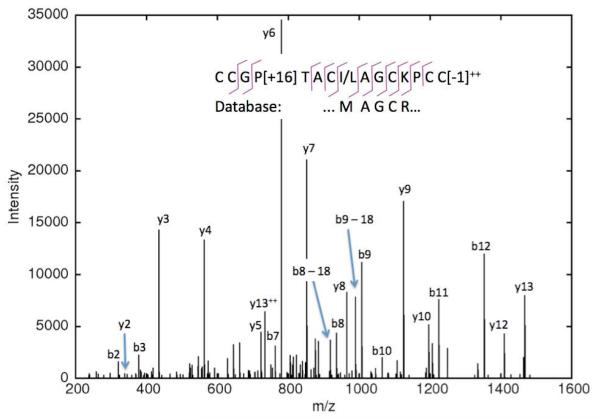
De novo peptide sequencing by mass spectrometry (MS) can determine the amino acid sequence of an unknown peptide without reference to a protein database. MS-based de novo sequencing assumes special importance in focused studies of families of biologically active peptides and proteins, such as hormones, toxins, and antibodies, for which amino acid sequences may be difficult to obtain through genomic methods. These protein families often exhibit sequence homology or characteristic amino acid content; yet, current de novo sequencing approaches do not take advantage of this prior knowledge and, hence, search an unnecessarily large space of possible sequences. Here, we describe an algorithm for de novo sequencing that incorporates sequence constraints into the core graph algorithm and thereby reduces the search space by many orders of magnitude. We demonstrate our algorithm in a study of cysteine-rich toxins from two cone snail species (Conus textile and Conus stercusmuscarum) and report 13 de novo and about 60 total toxins.
通过质谱(MS)从头测序肽可以确定未知肽的氨基酸序列,而无需参考蛋白质数据库。基于 MS 的从头测序在针对生物活性肽和蛋白质家族的重点研究中特别重要,例如激素、毒素和抗体,通过基因组方法获得这些家族的氨基酸序列可能很困难。这些蛋白质家族通常表现出序列同源性或特征性氨基酸含量;然而,目前的从头测序方法没有利用这种先验知识,因此搜索了不必要的大量可能序列空间。在这里,我们描述了一种将序列约束纳入核心图算法的从头测序算法,从而将搜索空间缩小了许多数量级。我们在对来自两种圆锥蜗牛物种(Conus textile 和 Conus stercusmuscarum)的富含半胱氨酸的毒素的研究中演示了我们的算法,并报告了 13 种从头和大约 60 种总毒素。