School of Medicine-Pathology, Clinical Science Institute, National University of Ireland Galway, Galway, Ireland.
Cell Death Dis. 2012 Jun 28;3(6):e333. doi: 10.1038/cddis.2012.74.
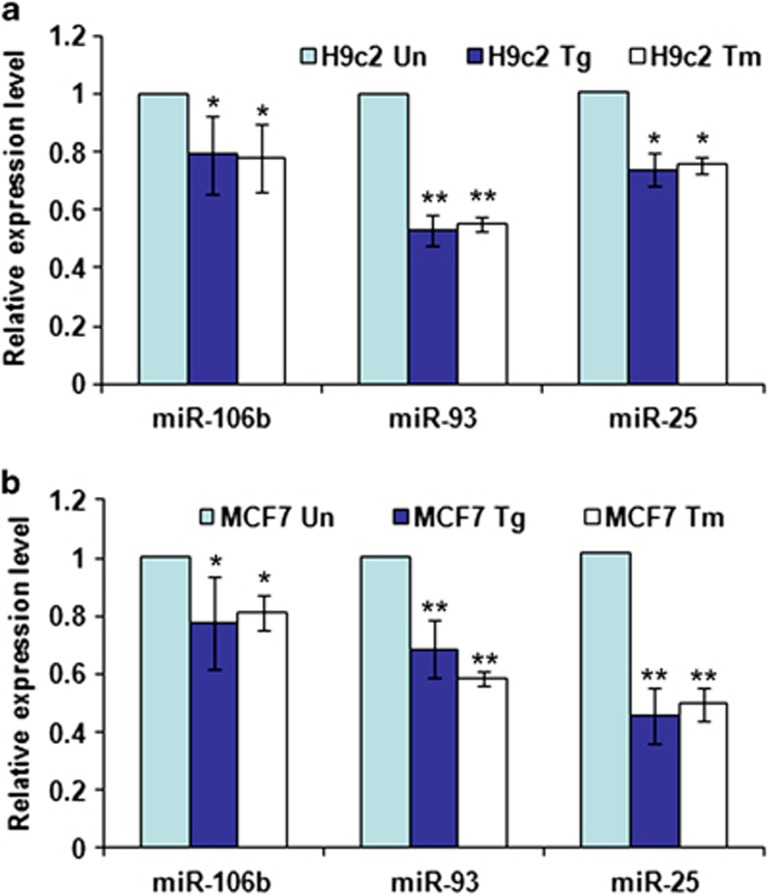
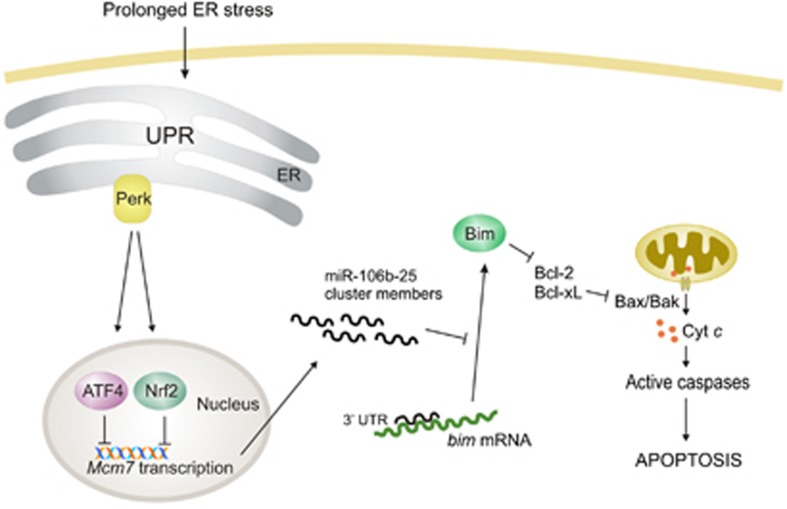
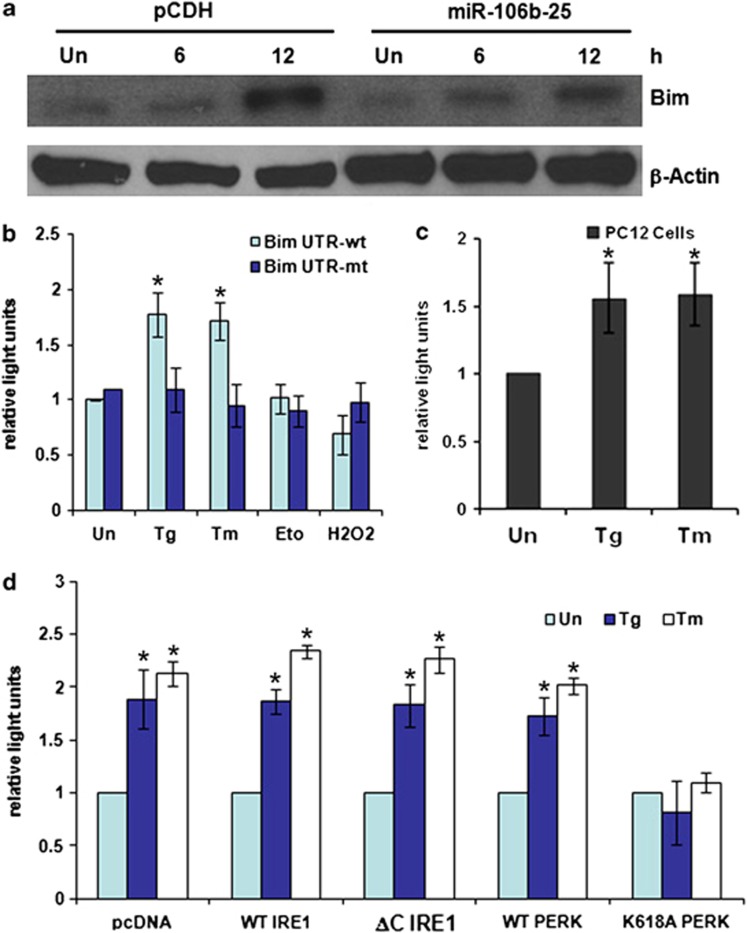
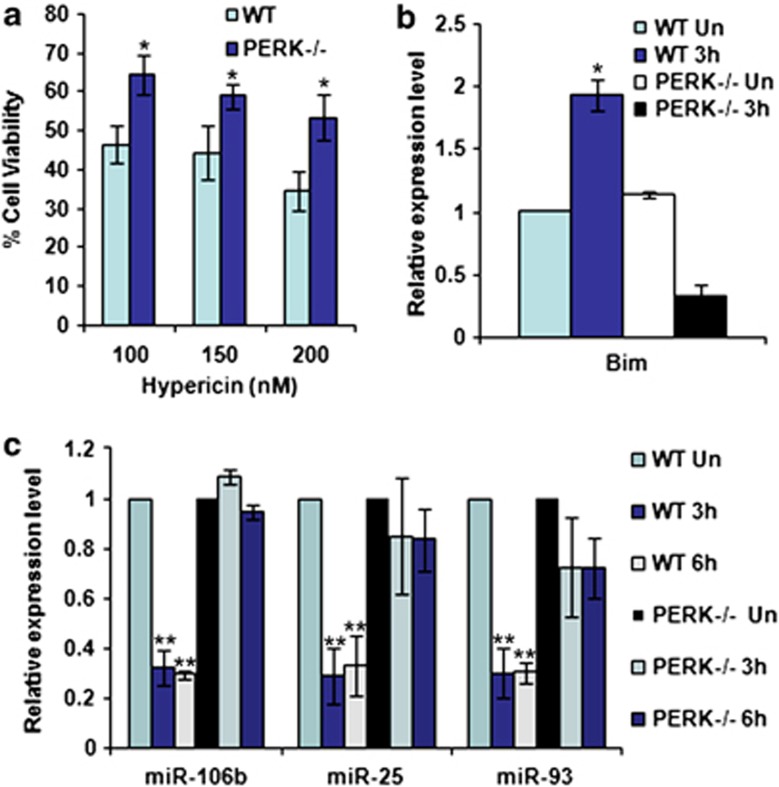
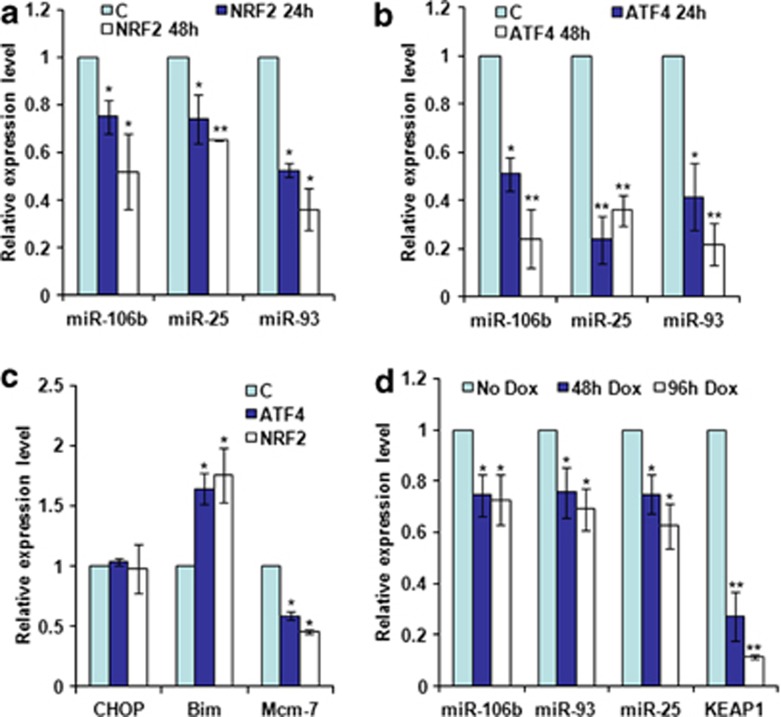
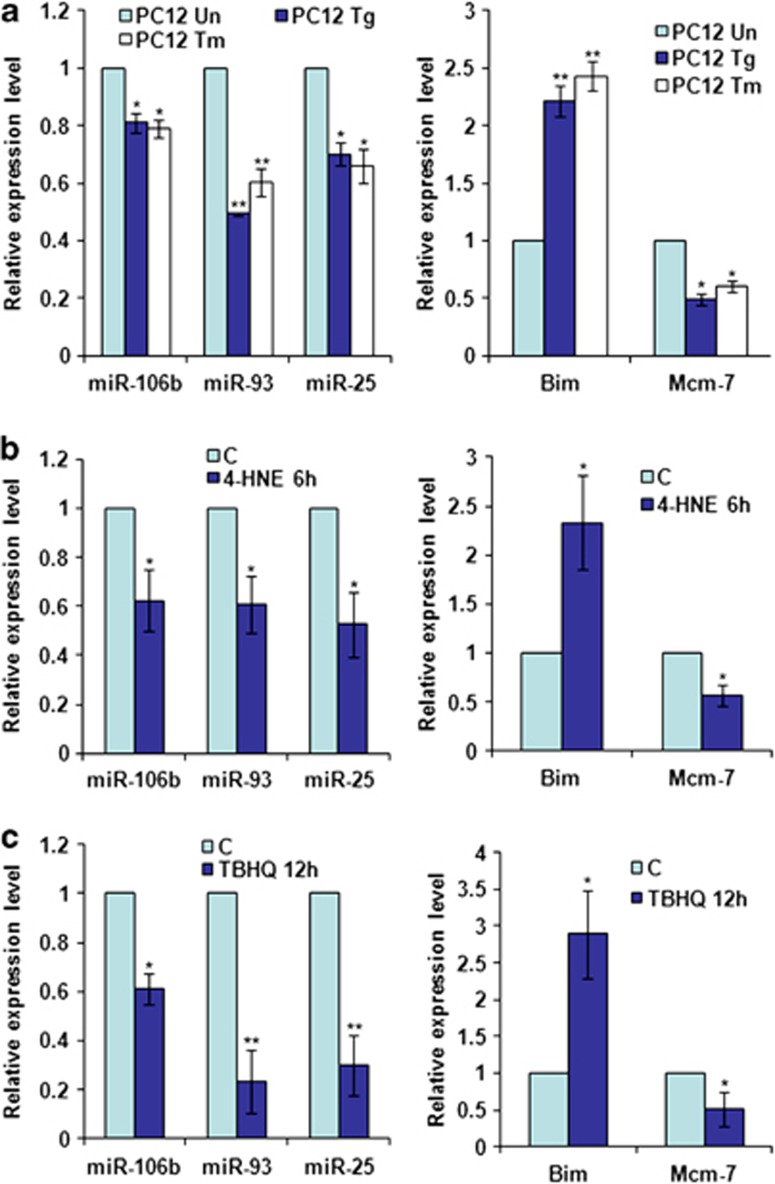
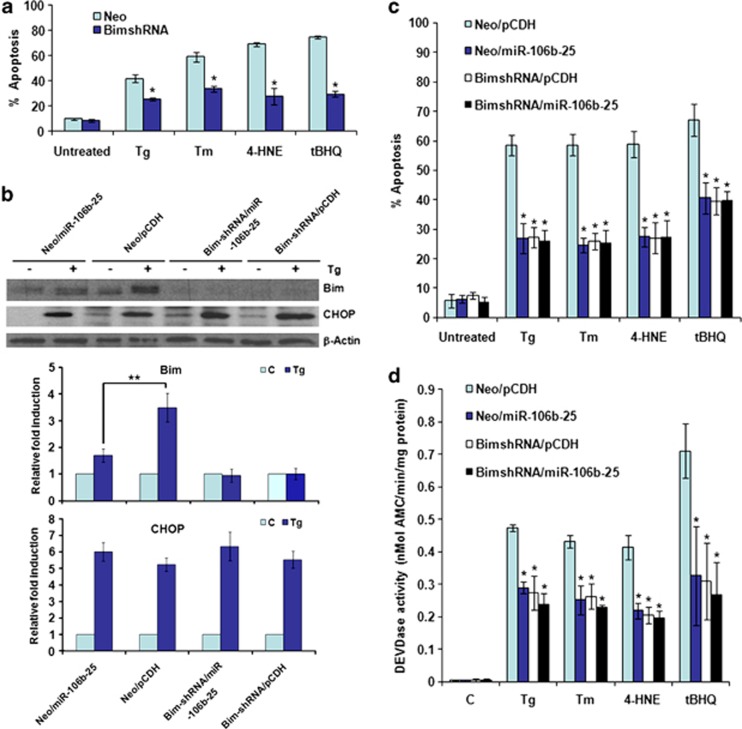
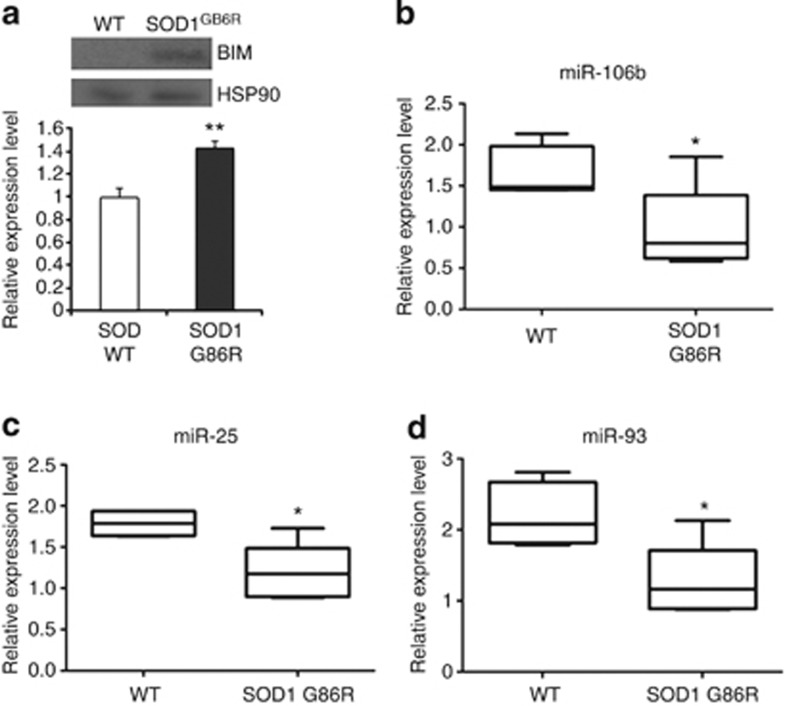
Activation of the unfolded protein response sensor PKR-like endoplasmic reticulum kinase (Perk) attenuates endoplasmic reticulum (ER) stress levels. Conversantly, if the damage is too severe and ER function cannot be restored, this signaling branch triggers apoptosis. Bcl-2 homology 3-only family member Bim is essential for ER stress-induced apoptosis. However, the regulatory mechanisms controlling Bim activation under ER stress conditions are not well understood. Here, we show that downregulation of the miR-106b-25 cluster contributes to ER stress-induced apoptosis and the upregulation of Bim. Hypericin-mediated photo-oxidative ER damage induced Perk-dependent cell death and led to a significant decrease in the levels of miRNAs belonging to miR-106b-25 cluster in wild-type (WT) but not in Perk⁻/⁻ MEFs. Further, we show that expression of miR-106b-25 and Mcm-7 (host gene of miR-106b-25) is co-regulated through the transcription factors Atf4 (activating transcription factor 4) and Nrf2 (nuclear factor-erythroid-2-related factor 2). ER stress increased the activity of WT Bim 3'UTR (untranslated region) construct but not the miR-106b-25 recognition site-mutated Bim 3'UTR construct. Overexpression of miR-106b-25 cluster inhibits ER stress-induced cell death in WT but did not confer any further protection in Bim-knockdown cells. Further, we show downregulation in the levels of miR-106b-25 cluster in the symptomatic SOD1(G86R) transgenic mice. Our results suggest a molecular mechanism whereby repression of miR-106b-25 cluster has an important role in ER stress-mediated increase in Bim and apoptosis.
未折叠蛋白反应传感器 PKR 样内质网激酶(Perk)的激活可减轻内质网(ER)应激水平。相反,如果损伤过于严重,内质网功能无法恢复,该信号分支则会触发细胞凋亡。Bcl-2 同源结构域 3 仅有家族成员 Bim 是 ER 应激诱导细胞凋亡所必需的。然而,在 ER 应激条件下控制 Bim 激活的调节机制尚不清楚。在这里,我们表明 miR-106b-25 簇的下调有助于 ER 应激诱导的细胞凋亡和 Bim 的上调。金丝桃素介导的光氧化内质网损伤诱导 Perk 依赖性细胞死亡,并导致野生型(WT)而非 Perk⁻/⁻ MEFs 中属于 miR-106b-25 簇的 miRNAs 水平显著降低。此外,我们表明 miR-106b-25 和 Mcm-7(miR-106b-25 的宿主基因)的表达通过转录因子 Atf4(激活转录因子 4)和 Nrf2(核因子-红细胞 2 相关因子 2)共同调节。ER 应激增加了 WT Bim 3'UTR(非翻译区)构建体的活性,但不增加 miR-106b-25 识别位点突变的 Bim 3'UTR 构建体的活性。miR-106b-25 簇的过表达抑制了 WT 细胞中的 ER 应激诱导的细胞死亡,但在 Bim 敲低细胞中没有提供任何进一步的保护。此外,我们还发现症状性 SOD1(G86R)转基因小鼠中 miR-106b-25 簇的水平下调。我们的研究结果表明了一种分子机制,即 miR-106b-25 簇的抑制在 ER 应激介导的 Bim 和细胞凋亡增加中起着重要作用。