Department of Computer Science and Engineering, Lehigh University, Bethlehem, PA, USA.
Proteome Sci. 2012 Jun 21;10 Suppl 1(Suppl 1):S6. doi: 10.1186/1477-5956-10-S1-S6.
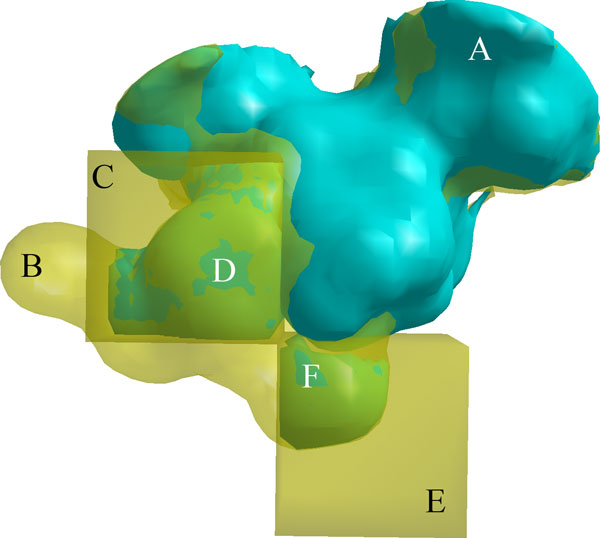
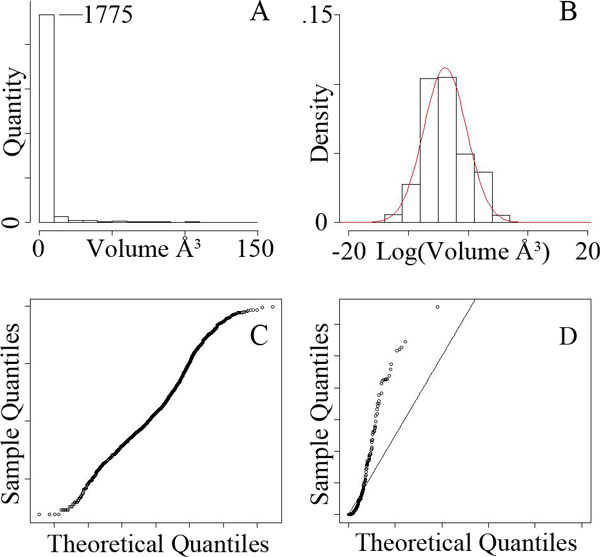
Identifying elements of protein structures that create differences in protein-ligand binding specificity is an essential method for explaining the molecular mechanisms underlying preferential binding. In some cases, influential mechanisms can be visually identified by experts in structural biology, but subtler mechanisms, whose significance may only be apparent from the analysis of many structures, are harder to find. To assist this process, we present a geometric algorithm and two statistical models for identifying significant structural differences in protein-ligand binding cavities. We demonstrate these methods in an analysis of sequentially nonredundant structural representatives of the canonical serine proteases and the enolase superfamily. Here, we observed that statistically significant structural variations identified experimentally established determinants of specificity. We also observed that an analysis of individual regions inside cavities can reveal areas where small differences in shape can correspond to differences in specificity.
确定导致蛋白质-配体结合特异性差异的蛋白质结构元素是解释优先结合的分子机制的重要方法。在某些情况下,结构生物学专家可以通过视觉识别有影响力的机制,但更微妙的机制,其意义可能只有通过对许多结构的分析才能显现出来,因此更难发现。为了辅助这一过程,我们提出了一种几何算法和两种统计模型,用于识别蛋白质-配体结合腔中的显著结构差异。我们在对经典丝氨酸蛋白酶和烯醇酶超家族的顺序非冗余结构代表进行分析时演示了这些方法。在这里,我们观察到实验确定的特异性决定因素的统计显著结构变化。我们还观察到,对腔体内各个区域的分析可以揭示形状上的微小差异可能对应于特异性差异的区域。