Chang Chun-Tien, Tsai Chi-Neu, Tang Chuan Yi, Chen Chun-Houh, Lian Jang-Hau, Hu Chi-Yu, Tsai Chia-Lung, Chao Angel, Lai Chyong-Huey, Wang Tzu-Hao, Lee Yun-Shien
Department of Computer Science, National Tsing Hua University, Hsin-Chu, Taiwan.
ScientificWorldJournal. 2012;2012:365104. doi: 10.1100/2012/365104. Epub 2012 Jun 18.
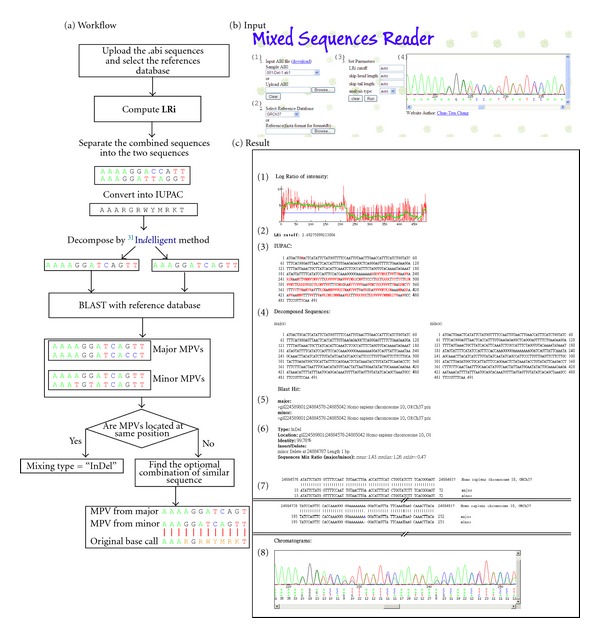
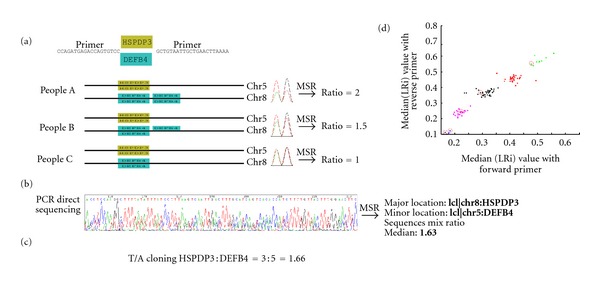
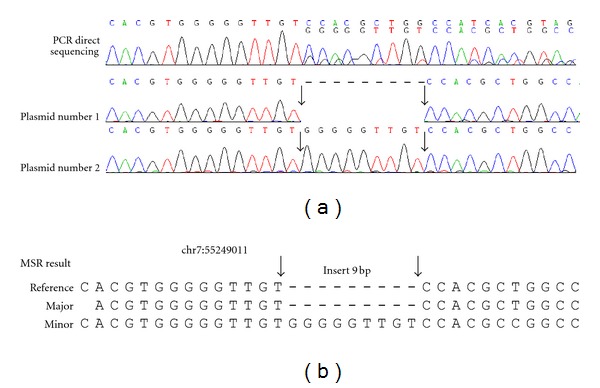
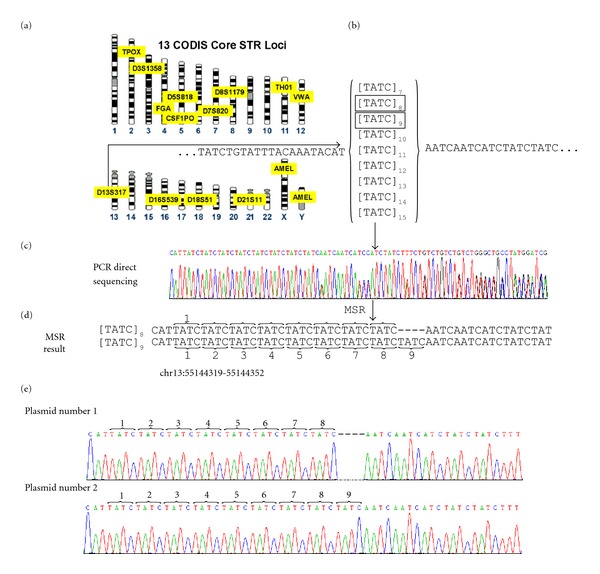
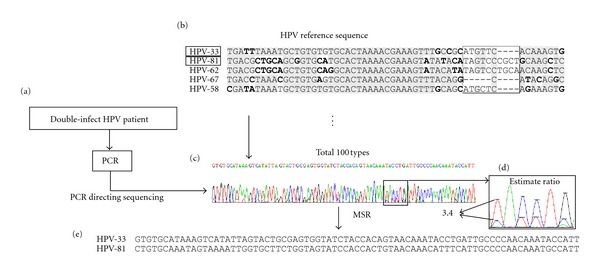
The direct sequencing of PCR products generates heterozygous base-calling fluorescence chromatograms that are useful for identifying single-nucleotide polymorphisms (SNPs), insertion-deletions (indels), short tandem repeats (STRs), and paralogous genes. Indels and STRs can be easily detected using the currently available Indelligent or ShiftDetector programs, which do not search reference sequences. However, the detection of other genomic variants remains a challenge due to the lack of appropriate tools for heterozygous base-calling fluorescence chromatogram data analysis. In this study, we developed a free web-based program, Mixed Sequence Reader (MSR), which can directly analyze heterozygous base-calling fluorescence chromatogram data in .abi file format using comparisons with reference sequences. The heterozygous sequences are identified as two distinct sequences and aligned with reference sequences. Our results showed that MSR may be used to (i) physically locate indel and STR sequences and determine STR copy number by searching NCBI reference sequences; (ii) predict combinations of microsatellite patterns using the Federal Bureau of Investigation Combined DNA Index System (CODIS); (iii) determine human papilloma virus (HPV) genotypes by searching current viral databases in cases of double infections; (iv) estimate the copy number of paralogous genes, such as β-defensin 4 (DEFB4) and its paralog HSPDP3.
对聚合酶链式反应(PCR)产物进行直接测序可生成杂合碱基呼叫荧光色谱图,这些色谱图有助于识别单核苷酸多态性(SNP)、插入缺失(indel)、短串联重复序列(STR)和旁系同源基因。使用当前可用的无需搜索参考序列的Indelligent或ShiftDetector程序可以轻松检测indel和STR。然而,由于缺乏用于杂合碱基呼叫荧光色谱图数据分析的合适工具,检测其他基因组变异仍然是一项挑战。在本研究中,我们开发了一个基于网络的免费程序,混合序列阅读器(MSR),它可以通过与参考序列进行比较,直接分析.abi文件格式的杂合碱基呼叫荧光色谱图数据。杂合序列被识别为两个不同的序列,并与参考序列进行比对。我们的结果表明,MSR可用于:(i)通过搜索美国国立医学图书馆(NCBI)参考序列在物理上定位indel和STR序列,并确定STR拷贝数;(ii)使用联邦调查局联合DNA索引系统(CODIS)预测微卫星模式的组合;(iii)在双重感染的情况下,通过搜索当前病毒数据库来确定人乳头瘤病毒(HPV)基因型;(iv)估计旁系同源基因的拷贝数,如β-防御素4(DEFB4)及其旁系同源基因HSPDP3。