Department of Chemical and Environmental Engineering, Yale University, New Haven, CT 06511, USA.
BMC Bioinformatics. 2012 Jul 18;13:170. doi: 10.1186/1471-2105-13-170.
The k-mer hash length is a key factor affecting the output of de novo transcriptome assembly packages using de Bruijn graph algorithms. Assemblies constructed with varying single k-mer choices might result in the loss of unique contiguous sequences (contigs) and relevant biological information. A common solution to this problem is the clustering of single k-mer assemblies. Even though annotation is one of the primary goals of a transcriptome assembly, the success of assembly strategies does not consider the impact of k-mer selection on the annotation output. This study provides an in-depth k-mer selection analysis that is focused on the degree of functional annotation achieved for a non-model organism where no reference genome information is available. Individual k-mers and clustered assemblies (CA) were considered using three representative software packages. Pair-wise comparison analyses (between individual k-mers and CAs) were produced to reveal missing Kyoto Encyclopedia of Genes and Genomes (KEGG) ortholog identifiers (KOIs), and to determine a strategy that maximizes the recovery of biological information in a de novo transcriptome assembly.
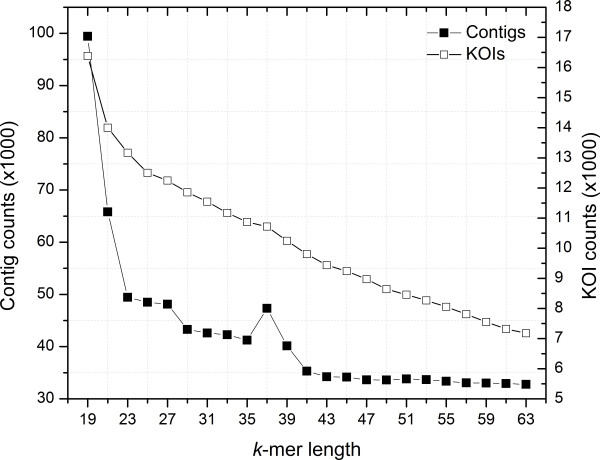
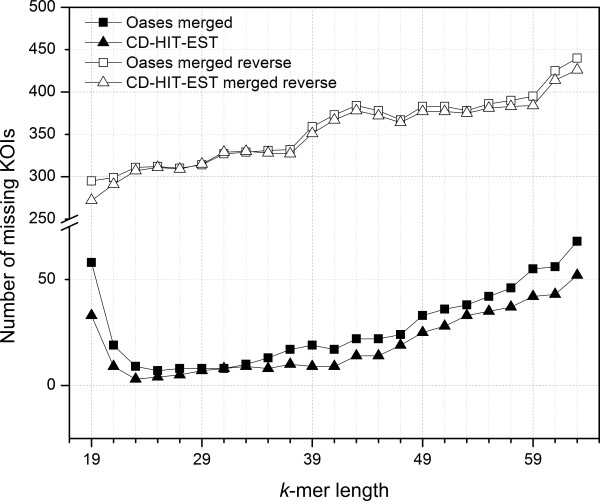
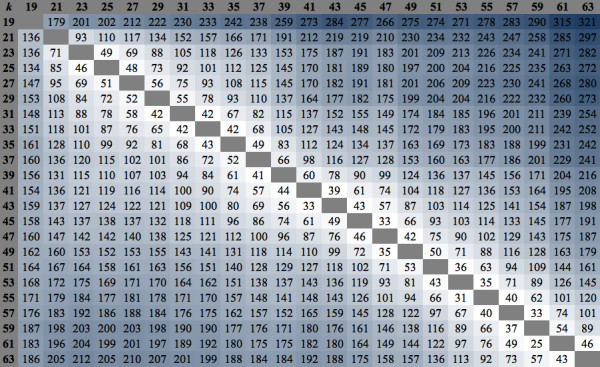
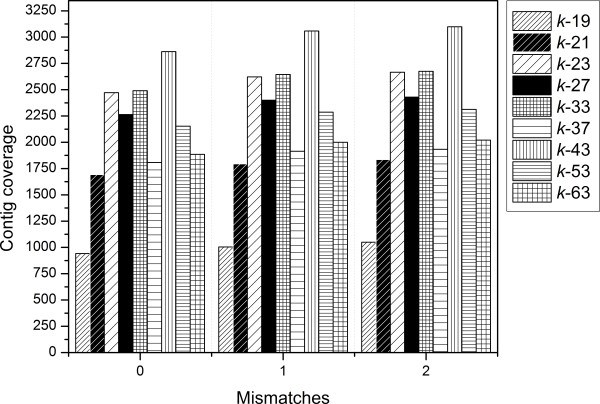
Analyses of single k-mer assemblies resulted in the generation of various quantities of contigs and functional annotations within the selection window of k-mers (k-19 to k-63). For each k-mer in this window, generated assemblies contained certain unique contigs and KOIs that were not present in the other k-mer assemblies. Producing a non-redundant CA of k-mers 19 to 63 resulted in a more complete functional annotation than any single k-mer assembly. However, a fraction of unique annotations remained (~0.19 to 0.27% of total KOIs) in the assemblies of individual k-mers (k-19 to k-63) that were not present in the non-redundant CA. A workflow to recover these unique annotations is presented.
This study demonstrated that different k-mer choices result in various quantities of unique contigs per single k-mer assembly which affects biological information that is retrievable from the transcriptome. This undesirable effect can be minimized, but not eliminated, with clustering of multi-k assemblies with redundancy removal. The complete extraction of biological information in de novo transcriptomics studies requires both the production of a CA and efforts to identify unique contigs that are present in individual k-mer assemblies but not in the CA.
在使用 de Bruijn 图算法的从头转录组组装包中,k-mer 哈希长度是影响输出的关键因素。使用不同的单个 k-mer 选择构建的组装可能会导致独特的连续序列(contigs)和相关的生物信息丢失。解决此问题的常用方法是对单个 k-mer 组装进行聚类。尽管注释是转录组组装的主要目标之一,但组装策略的成功与否并未考虑 k-mer 选择对注释输出的影响。本研究提供了一种深入的 k-mer 选择分析,重点关注在没有参考基因组信息的情况下,针对非模式生物达到的功能注释程度。使用三个有代表性的软件包考虑了单个 k-mer 和聚类组装(CA)。产生了成对比较分析(在单个 k-mer 和 CA 之间),以揭示缺少京都基因与基因组百科全书(KEGG)直系同源标识符(KOI),并确定一种策略,使从头转录组组装中生物信息的恢复最大化。
单个 k-mer 组装的分析导致在 k-mer(k-19 到 k-63)选择窗口内生成了各种数量的 contigs 和功能注释。在该窗口中的每个 k-mer 中,生成的组装都包含某些在其他 k-mer 组装中不存在的独特 contigs 和 KOI。生成 k-19 到 63 的非冗余 k-mer CA 导致的功能注释比任何单个 k-mer 组装都更完整。然而,在不在非冗余 CA 中的单个 k-mer(k-19 到 k-63)组装中仍然存在一些独特的注释(占总 KOI 的 0.19 到 0.27%)。提出了一种恢复这些独特注释的工作流程。
本研究表明,不同的 k-mer 选择会导致每个单个 k-mer 组装中独特 contigs 的数量不同,从而影响可从转录组中检索到的生物信息。通过去除冗余的多 k 组装聚类,可以最小化但不能消除这种不良影响。在从头转录组学研究中提取完整的生物信息既需要生成 CA,又需要努力识别存在于单个 k-mer 组装中但不存在于 CA 中的独特 contigs。