Biodiversity and Climate Research Center (BiK-F), Molecular Ecology Group, Biocampus Siesmayerstraße, Goethe-Universität, Frankfurt am Main, Germany.
BMC Genomics. 2011 Jun 16;12:317. doi: 10.1186/1471-2164-12-317.
Until recently, read lengths on the Solexa/Illumina system were too short to reliably assemble transcriptomes without a reference sequence, especially for non-model organisms. However, with read lengths up to 100 nucleotides available in the current version, an assembly without reference genome should be possible. For this study we created an EST data set for the common pond snail Radix balthica by Illumina sequencing of a normalized transcriptome. Performance of three different short read assemblers was compared with respect to: the number of contigs, their length, depth of coverage, their quality in various BLAST searches and the alignment to mitochondrial genes.
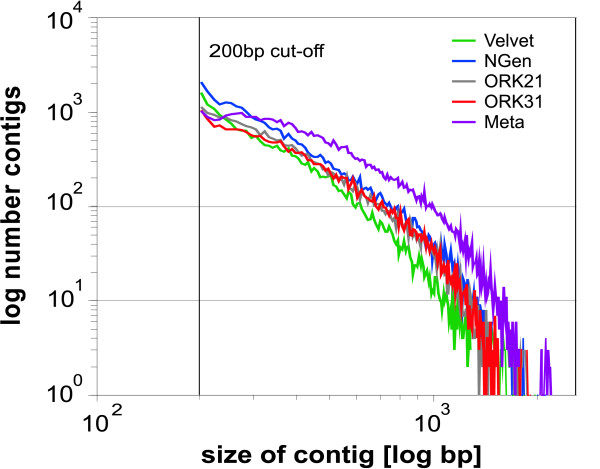
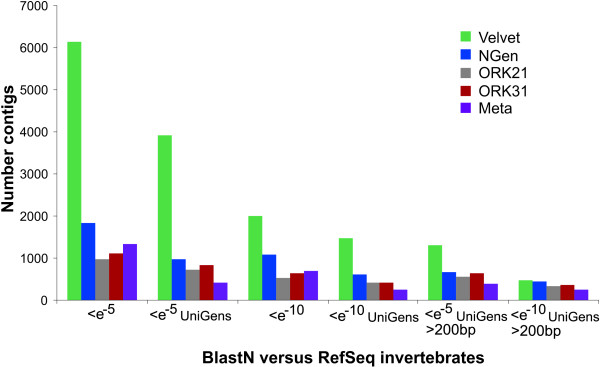
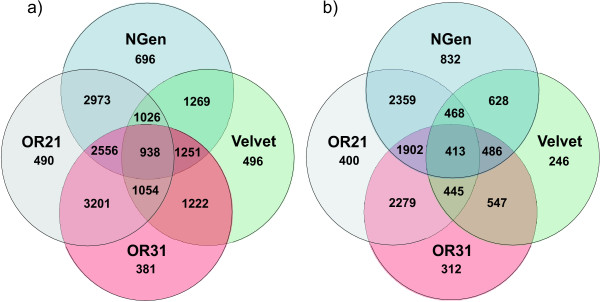
A single sequencing run of a normalized RNA pool resulted in 16,923,850 paired end reads with median read length of 61 bases. The assemblies generated by VELVET, OASES, and SeqMan NGEN differed in the total number of contigs, contig length, the number and quality of gene hits obtained by BLAST searches against various databases, and contig performance in the mt genome comparison. While VELVET produced the highest overall number of contigs, a large fraction of these were of small size (< 200bp), and gave redundant hits in BLAST searches and the mt genome alignment. The best overall contig performance resulted from the NGEN assembly. It produced the second largest number of contigs, which on average were comparable to the OASES contigs but gave the highest number of gene hits in two out of four BLAST searches against different reference databases. A subsequent meta-assembly of the four contig sets resulted in larger contigs, less redundancy and a higher number of BLAST hits.
Our results document the first de novo transcriptome assembly of a non-model species using Illumina sequencing data. We show that de novo transcriptome assembly using this approach yields results useful for downstream applications, in particular if a meta-assembly of contig sets is used to increase contig quality. These results highlight the ongoing need for improvements in assembly methodology.
直到最近,Solexa/Illumina 系统的读取长度还太短,无法在没有参考序列的情况下可靠地组装转录组,尤其是对于非模式生物。然而,当前版本的读取长度可达 100 个核苷酸,因此应该有可能在没有参考基因组的情况下进行组装。在这项研究中,我们通过 Illumina 对标准化转录组进行测序,为常见的池塘蜗牛 Radix balthica 创建了一个 EST 数据集。我们比较了三种不同的短读序列组装程序的性能,具体涉及:contigs 的数量、长度、覆盖深度、在各种 BLAST 搜索中的质量以及与线粒体基因的比对。
单个标准化 RNA 池的测序运行产生了 16,923,850 对末端读取,中位数读取长度为 61 个碱基。VELVET、OASES 和 SeqMan NGEN 生成的组装在 contigs 的总数、contig 长度、通过针对各种数据库的 BLAST 搜索获得的基因命中数量和质量以及在 mt 基因组比较中的 contig 性能方面存在差异。虽然 VELVET 产生了最高数量的 contigs,但其中很大一部分是小尺寸的(<200bp),并且在 BLAST 搜索和 mt 基因组比对中产生了冗余的命中。来自 NGEN 组装的整体最佳 contig 性能。它产生了第二多的 contigs,平均而言与 OASES 的 contigs 相当,但在针对四个不同参考数据库中的两个 BLAST 搜索中产生了最多的基因命中。随后对四个 contig 集进行的元组装产生了更大的 contigs,更少的冗余和更多的 BLAST 命中。
我们的结果记录了首次使用 Illumina 测序数据对非模式物种进行从头转录组组装。我们表明,使用这种方法进行从头转录组组装可产生可用于下游应用的结果,特别是如果使用 contig 集的元组装来提高 contig 质量。这些结果突出表明组装方法学需要不断改进。