Division of Pediatric Neurooncology, German Cancer Research Center (DKFZ), Im Neuenheimer Feld 280, Heidelberg 69120, Germany.
Nature. 2012 Aug 2;488(7409):100-5. doi: 10.1038/nature11284.
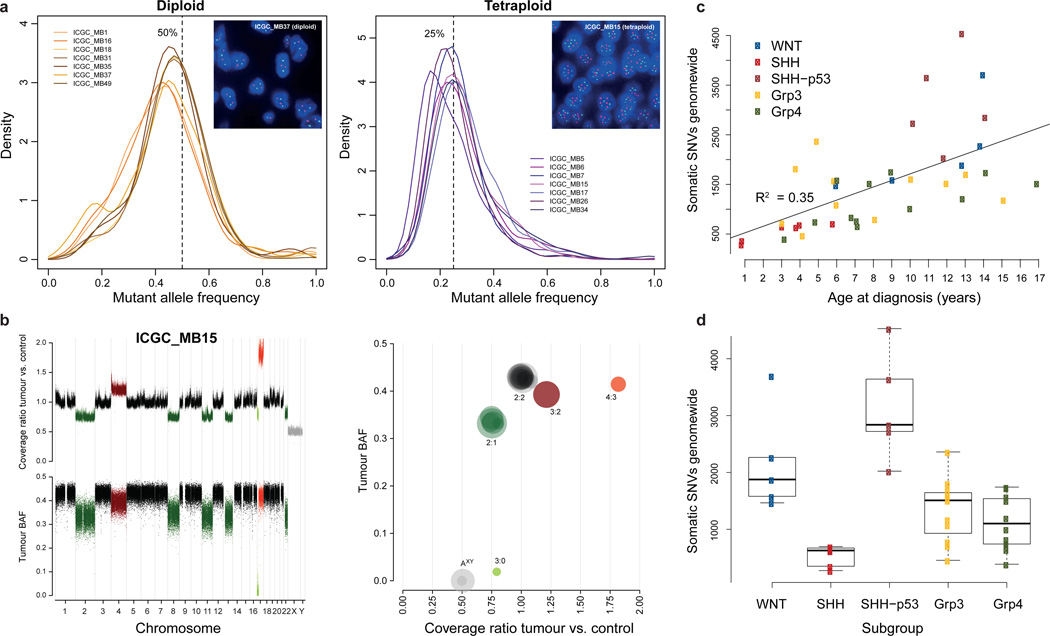
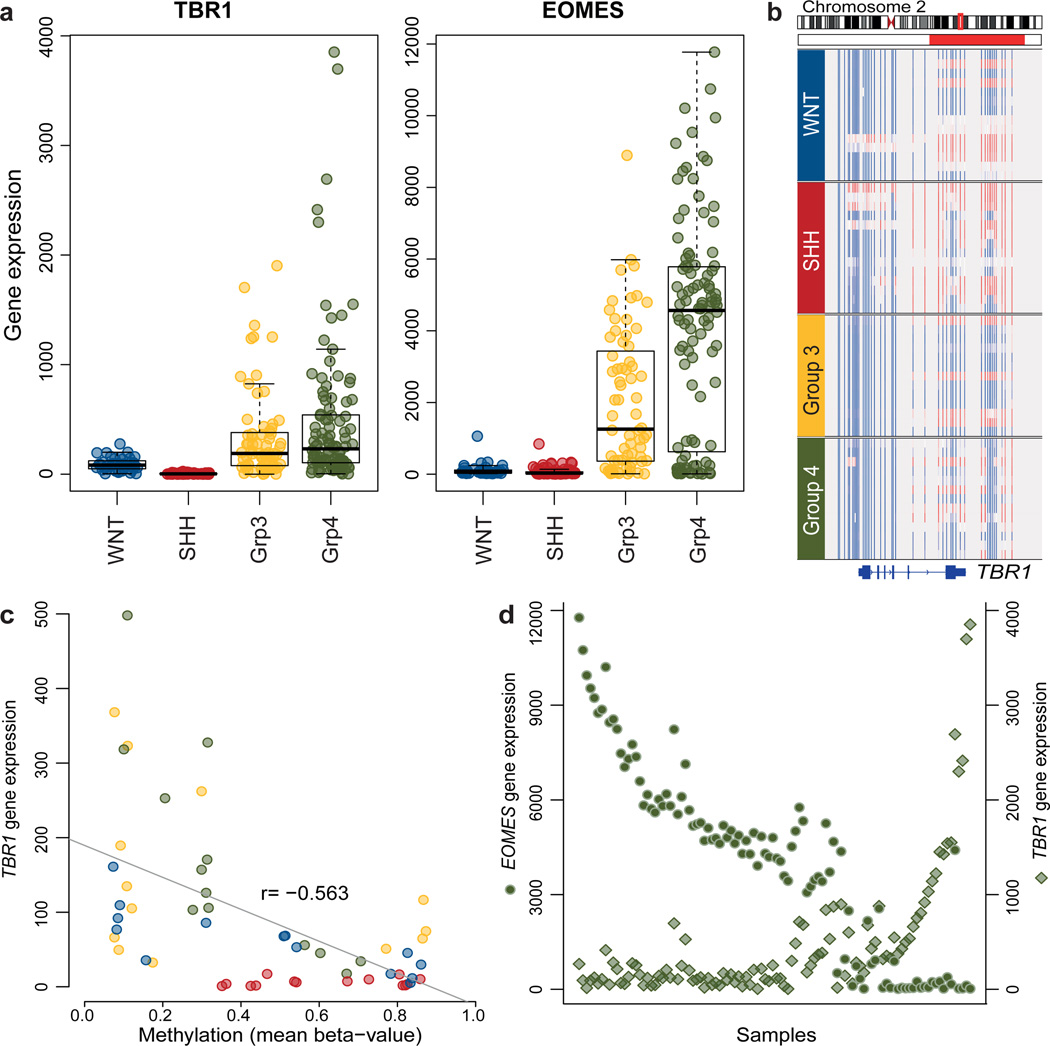
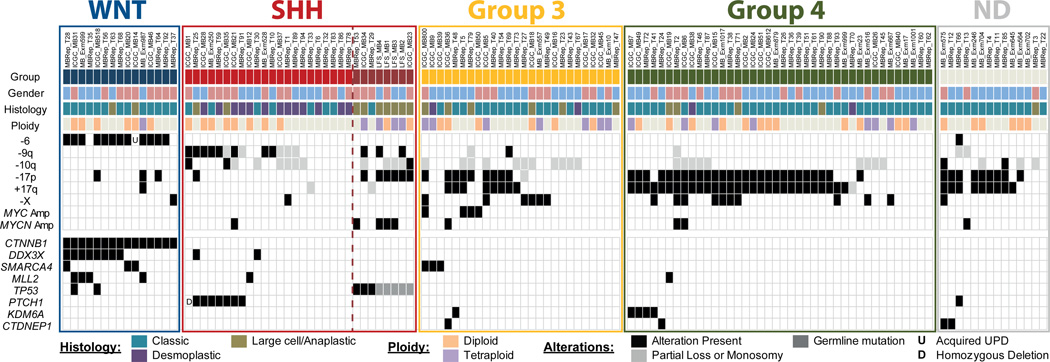
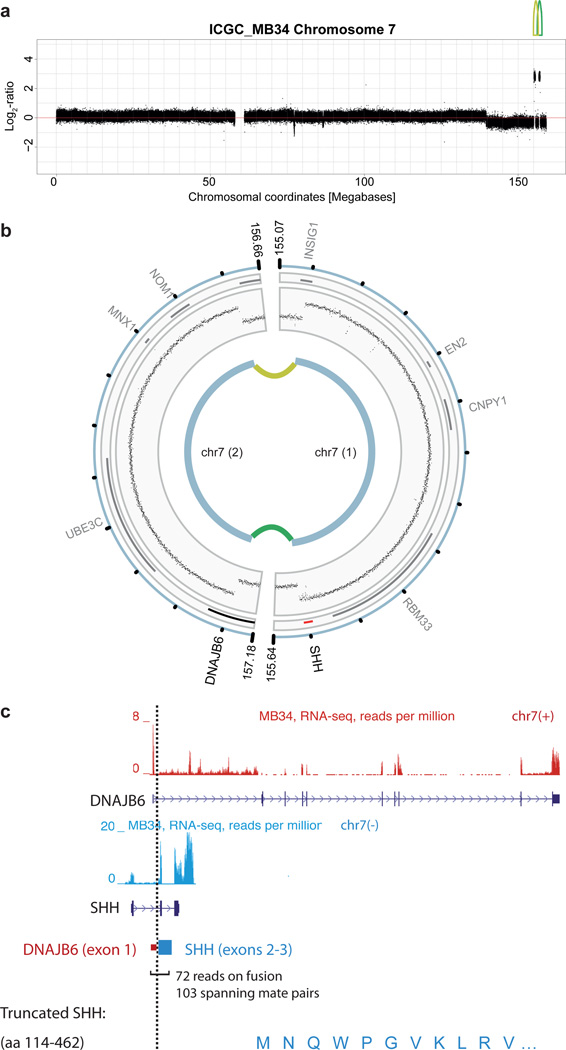
Medulloblastoma is an aggressively growing tumour, arising in the cerebellum or medulla/brain stem. It is the most common malignant brain tumour in children, and shows tremendous biological and clinical heterogeneity. Despite recent treatment advances, approximately 40% of children experience tumour recurrence, and 30% will die from their disease. Those who survive often have a significantly reduced quality of life. Four tumour subgroups with distinct clinical, biological and genetic profiles are currently identified. WNT tumours, showing activated wingless pathway signalling, carry a favourable prognosis under current treatment regimens. SHH tumours show hedgehog pathway activation, and have an intermediate prognosis. Group 3 and 4 tumours are molecularly less well characterized, and also present the greatest clinical challenges. The full repertoire of genetic events driving this distinction, however, remains unclear. Here we describe an integrative deep-sequencing analysis of 125 tumour-normal pairs, conducted as part of the International Cancer Genome Consortium (ICGC) PedBrain Tumor Project. Tetraploidy was identified as a frequent early event in Group 3 and 4 tumours, and a positive correlation between patient age and mutation rate was observed. Several recurrent mutations were identified, both in known medulloblastoma-related genes (CTNNB1, PTCH1, MLL2, SMARCA4) and in genes not previously linked to this tumour (DDX3X, CTDNEP1, KDM6A, TBR1), often in subgroup-specific patterns. RNA sequencing confirmed these alterations, and revealed the expression of what are, to our knowledge, the first medulloblastoma fusion genes identified. Chromatin modifiers were frequently altered across all subgroups. These findings enhance our understanding of the genomic complexity and heterogeneity underlying medulloblastoma, and provide several potential targets for new therapeutics, especially for Group 3 and 4 patients.
髓母细胞瘤是一种侵袭性生长的肿瘤,起源于小脑或延髓/脑干。它是儿童中最常见的恶性脑肿瘤,表现出巨大的生物学和临床异质性。尽管最近的治疗进展,大约 40%的儿童经历肿瘤复发,30%的儿童死于疾病。那些幸存下来的人通常生活质量显著降低。目前已经确定了四个具有不同临床、生物学和遗传特征的肿瘤亚组。WNT 肿瘤,表现出激活的无翅信号通路,在当前的治疗方案下预后良好。SHH 肿瘤表现出 Hedgehog 通路激活,具有中等预后。3 组和 4 组肿瘤在分子上的特征不太明显,也带来了最大的临床挑战。然而,驱动这种区别的遗传事件的全部范围仍然不清楚。在这里,我们描述了作为国际癌症基因组联盟(ICGC)PedBrain 肿瘤项目的一部分进行的 125 对肿瘤-正常对的综合深度测序分析。四倍体被确定为 3 组和 4 组肿瘤的常见早期事件,并且观察到患者年龄和突变率之间的正相关。鉴定了几个复发性突变,既有已知的髓母细胞瘤相关基因(CTNNB1、PTCH1、MLL2、SMARCA4)的突变,也有以前与这种肿瘤无关的基因(DDX3X、CTDNEP1、KDM6A、TBR1)的突变,通常具有亚组特异性模式。RNA 测序证实了这些改变,并揭示了我们所知的第一个髓母细胞瘤融合基因的表达。染色质修饰物在所有亚组中经常改变。这些发现增强了我们对髓母细胞瘤基础基因组复杂性和异质性的理解,并为新的治疗方法提供了几个潜在的靶点,特别是对于 3 组和 4 组患者。