Schwalbe Edward C, Lindsey Janet C, Nakjang Sirintra, Crosier Stephen, Smith Amanda J, Hicks Debbie, Rafiee Gholamreza, Hill Rebecca M, Iliasova Alice, Stone Thomas, Pizer Barry, Michalski Antony, Joshi Abhijit, Wharton Stephen B, Jacques Thomas S, Bailey Simon, Williamson Daniel, Clifford Steven C
Wolfson Childhood Cancer Research Centre, Northern Institute for Cancer Research, Newcastle University, Newcastle upon Tyne, UK; Department of Applied Sciences, Northumbria University, Newcastle upon Tyne, UK.
Wolfson Childhood Cancer Research Centre, Northern Institute for Cancer Research, Newcastle University, Newcastle upon Tyne, UK.
Lancet Oncol. 2017 Jul;18(7):958-971. doi: 10.1016/S1470-2045(17)30243-7. Epub 2017 May 22.
International consensus recognises four medulloblastoma molecular subgroups: WNT (MB), SHH (MB), group 3 (MB), and group 4 (MB), each defined by their characteristic genome-wide transcriptomic and DNA methylomic profiles. These subgroups have distinct clinicopathological and molecular features, and underpin current disease subclassification and initial subgroup-directed therapies that are underway in clinical trials. However, substantial biological heterogeneity and differences in survival are apparent within each subgroup, which remain to be resolved. We aimed to investigate whether additional molecular subgroups exist within childhood medulloblastoma and whether these could be used to improve disease subclassification and prognosis predictions.
In this retrospective cohort study, we assessed 428 primary medulloblastoma samples collected from UK Children's Cancer and Leukaemia Group (CCLG) treatment centres (UK), collaborating European institutions, and the UKCCSG-SIOP-PNET3 European clinical trial. An independent validation cohort (n=276) of archival tumour samples was also analysed. We analysed samples from patients with childhood medulloblastoma who were aged 0-16 years at diagnosis, and had central review of pathology and comprehensive clinical data. We did comprehensive molecular profiling, including DNA methylation microarray analysis, and did unsupervised class discovery of test and validation cohorts to identify consensus primary molecular subgroups and characterise their clinical and biological significance. We modelled survival of patients aged 3-16 years in patients (n=215) who had craniospinal irradiation and had been treated with a curative intent.
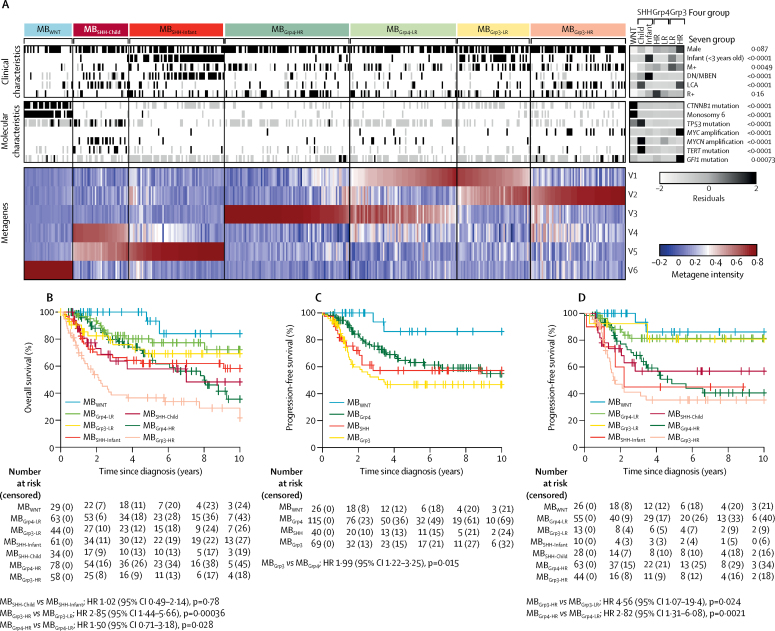
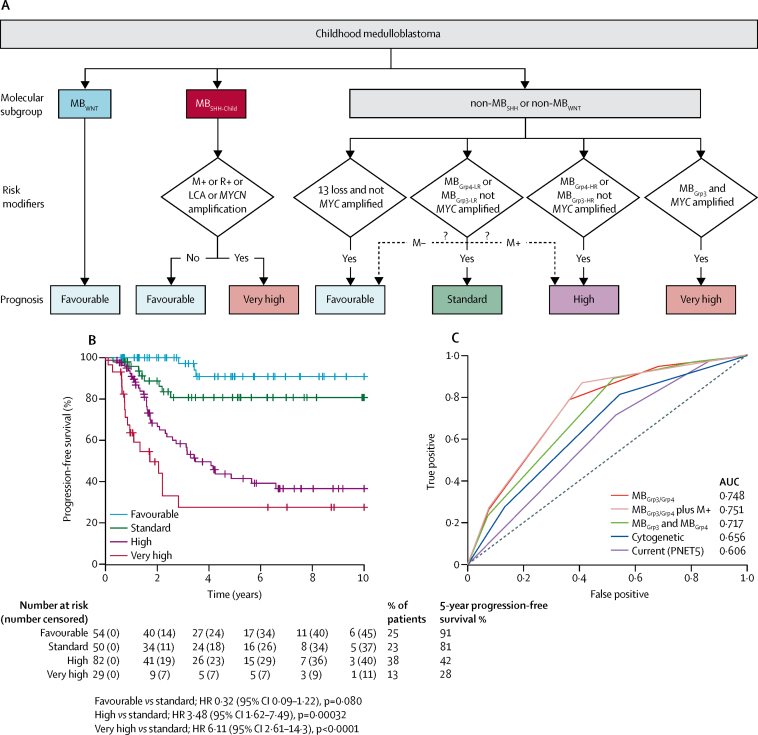
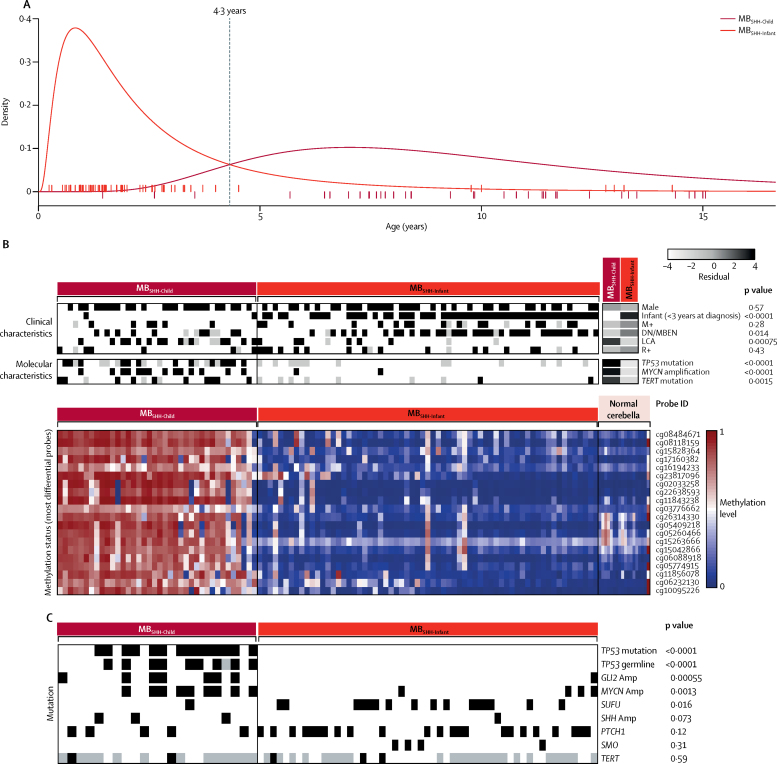
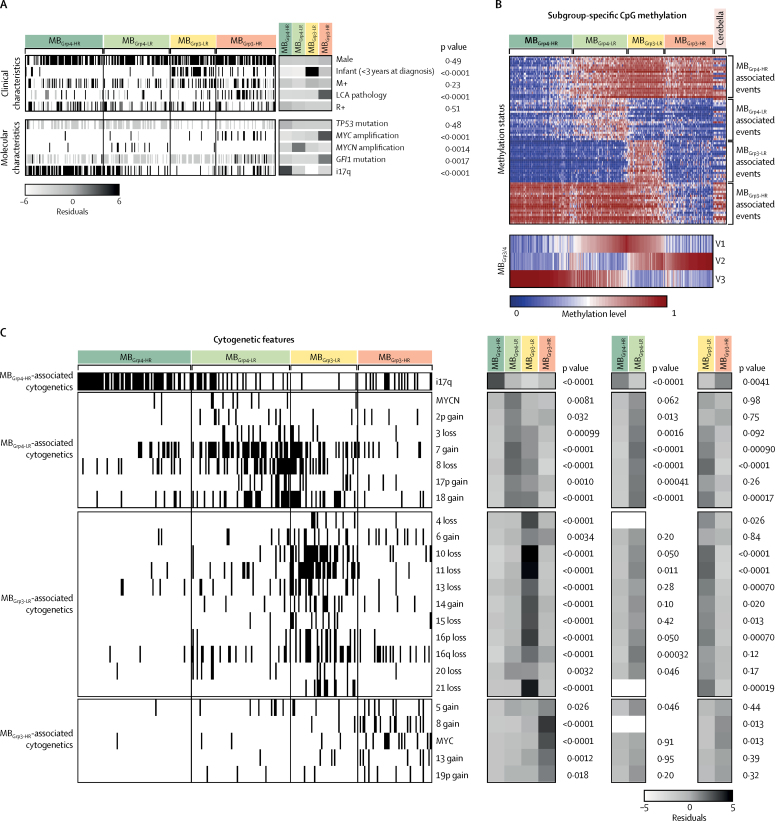
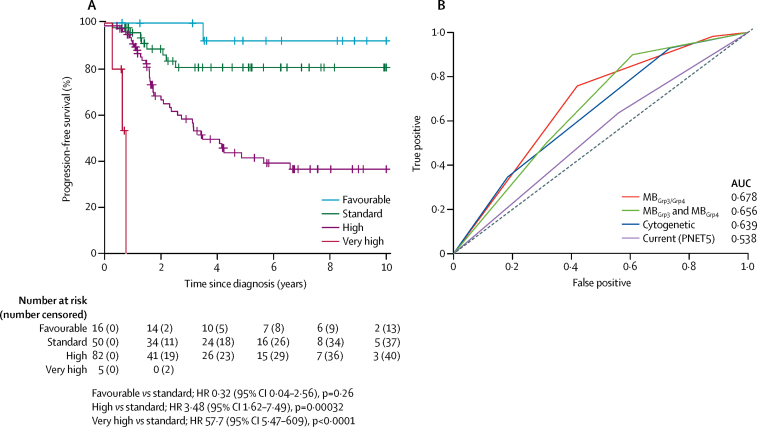
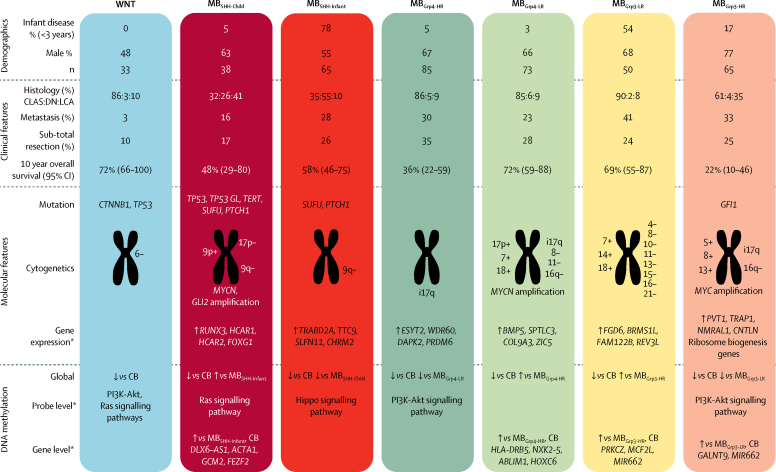
Seven robust and reproducible primary molecular subgroups of childhood medulloblastoma were identified. MB remained unchanged and each remaining consensus subgroup was split in two. MB was split into age-dependent subgroups corresponding to infant (<4·3 years; MB; n=65) and childhood patients (≥4·3 years; MB; n=38). MB and MB were each split into high-risk (MB [n=65] and MB [n=85]) and low-risk (MB [n=50] and MB [n=73]) subgroups. These biological subgroups were validated in the independent cohort. We identified features of the seven subgroups that were predictive of outcome. Cross-validated subgroup-dependent survival models, incorporating these novel subgroups along with secondary clinicopathological and molecular features and established disease risk-factors, outperformed existing disease risk-stratification schemes. These subgroup-dependent models stratified patients into four clinical risk groups for 5-year progression-free survival: favourable risk (54 [25%] of 215 patients; 91% survival [95% CI 82-100]); standard risk (50 [23%] patients; 81% survival [70-94]); high-risk (82 [38%] patients; 42% survival [31-56]); and very high-risk (29 [13%] patients; 28% survival [14-56]).
The discovery of seven novel, clinically significant subgroups improves disease risk-stratification and could inform treatment decisions. These data provide a new foundation for future research and clinical investigations.
Cancer Research UK, The Tom Grahame Trust, Star for Harris, Action Medical Research, SPARKS, The JGW Patterson Foundation, The INSTINCT network (co-funded by The Brain Tumour Charity, Great Ormond Street Children's Charity, and Children with Cancer UK).
国际共识认可四种髓母细胞瘤分子亚组:WNT(MB)、SHH(MB)、3组(MB)和4组(MB),每组均由其特征性的全基因组转录组和DNA甲基化组图谱定义。这些亚组具有不同的临床病理和分子特征,是当前疾病亚分类及正在进行的临床试验中初始亚组导向治疗的基础。然而,每个亚组内均存在明显的生物学异质性和生存差异,仍有待解决。我们旨在研究儿童髓母细胞瘤中是否存在其他分子亚组,以及这些亚组能否用于改善疾病亚分类和预后预测。
在这项回顾性队列研究中,我们评估了从英国儿童癌症与白血病研究组(CCLG)治疗中心(英国)、合作的欧洲机构以及UKCCSG-SIOP-PNET3欧洲临床试验收集的428份原发性髓母细胞瘤样本。还分析了一个独立的存档肿瘤样本验证队列(n = 276)。我们分析了诊断时年龄为0至16岁、经中心病理审查且有全面临床数据的儿童髓母细胞瘤患者的样本。我们进行了全面的分子谱分析,包括DNA甲基化微阵列分析,并对测试队列和验证队列进行无监督分类发现,以确定一致的原发性分子亚组,并表征其临床和生物学意义。我们对3至16岁接受了颅脊髓照射且接受了根治性治疗的患者(n = 215)的生存情况进行了建模。
确定了儿童髓母细胞瘤的七个稳健且可重复的原发性分子亚组。MB保持不变,其余每个一致亚组均一分为二。MB分为与婴儿(<4.3岁;MB;n = 65)和儿童患者(≥4.3岁;MB;n = 38)相对应的年龄依赖性亚组。MB和MB各自分为高危(MB [n = 65]和MB [n = 85])和低危(MB [n = 50]和MB [n = 73])亚组。这些生物学亚组在独立队列中得到了验证。我们确定了七个亚组中可预测预后的特征。纳入这些新亚组以及次要临床病理和分子特征及既定疾病风险因素的交叉验证亚组依赖性生存模型,优于现有的疾病风险分层方案。这些亚组依赖性模型将患者分为五年无进展生存的四个临床风险组:低危(215例患者中的54例[25%];生存率91% [95% CI 82 - 100]);中危(50例[23%]患者;生存率81% [70 - 94]);高危(82例[38%]患者;生存率42% [31 - 56]);以及极高危(29例[13%]患者;生存率28% [14 - 56])。
七个新的、具有临床意义的亚组的发现改善了疾病风险分层,并可为治疗决策提供参考。这些数据为未来的研究和临床调查提供了新的基础。
英国癌症研究中心、汤姆·格雷厄姆信托基金、哈里斯之星、医学研究行动组织、SPARKS、JGW帕特森基金会、INSTINCT网络(由脑肿瘤慈善机构、大奥蒙德街儿童医院慈善机构和英国儿童癌症协会共同资助)。