Institute for Quantitative Biomedical Sciences, Department of Genetics, Dartmouth Medical School, One Medical Center Dr,, Lebanon, NH 03756, USA.
BioData Min. 2012 Jul 28;5(1):9. doi: 10.1186/1756-0381-5-9.
It is increasingly clear that common human diseases have a complex genetic architecture characterized by both additive and nonadditive genetic effects. The goal of the present study was to determine whether patterns of both additive and nonadditive genetic associations aggregate in specific functional groups as defined by the Gene Ontology (GO).
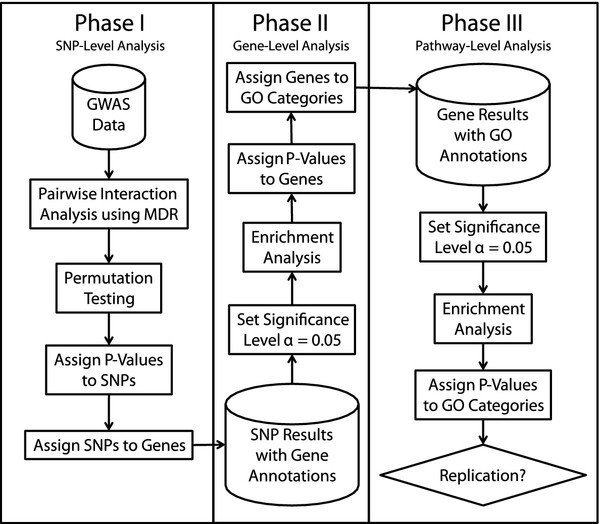
We first estimated all pairwise additive and nonadditive genetic effects using the multifactor dimensionality reduction (MDR) method that makes few assumptions about the underlying genetic model. Statistical significance was evaluated using permutation testing in two genome-wide association studies of ALS. The detection data consisted of 276 subjects with ALS and 271 healthy controls while the replication data consisted of 221 subjects with ALS and 211 healthy controls. Both studies included genotypes from approximately 550,000 single-nucleotide polymorphisms (SNPs). Each SNP was mapped to a gene if it was within 500 kb of the start or end. Each SNP was assigned a p-value based on its strongest joint effect with the other SNPs. We then used the Exploratory Visual Analysis (EVA) method and software to assign a p-value to each gene based on the overabundance of significant SNPs at the α = 0.05 level in the gene. We also used EVA to assign p-values to each GO group based on the overabundance of significant genes at the α = 0.05 level. A GO category was determined to replicate if that category was significant at the α = 0.05 level in both studies. We found two GO categories that replicated in both studies. The first, 'Regulation of Cellular Component Organization and Biogenesis', a GO Biological Process, had p-values of 0.010 and 0.014 in the detection and replication studies, respectively. The second, 'Actin Cytoskeleton', a GO Cellular Component, had p-values of 0.040 and 0.046 in the detection and replication studies, respectively.
Pathway analysis of pairwise genetic associations in two GWAS of sporadic ALS revealed a set of genes involved in cellular component organization and actin cytoskeleton, more specifically, that were not reported by prior GWAS. However, prior biological studies have implicated actin cytoskeleton in ALS and other motor neuron diseases. This study supports the idea that pathway-level analysis of GWAS data may discover important associations not revealed using conventional one-SNP-at-a-time approaches.
越来越明显的是,常见的人类疾病具有复杂的遗传结构,其特征是既有加性遗传效应,也有非加性遗传效应。本研究的目的是确定加性和非加性遗传关联模式是否会聚集在特定的功能组中,这些功能组由基因本体论(GO)定义。
我们首先使用多因子维度缩减(MDR)方法估计所有的加性和非加性遗传效应,该方法对潜在遗传模型的假设很少。在两项肌萎缩侧索硬化症(ALS)的全基因组关联研究中,使用置换检验评估统计显著性。检测数据包括 276 名 ALS 患者和 271 名健康对照者,而复制数据包括 221 名 ALS 患者和 211 名健康对照者。这两项研究都包含了大约 550,000 个单核苷酸多态性(SNP)的基因型。如果 SNP 在起始或结束点的 500kb 内,则将其映射到一个基因上。根据与其他 SNP 的最强联合效应,每个 SNP 都被赋予一个 p 值。然后,我们使用探索性可视化分析(EVA)方法和软件,根据基因中显著 SNP 的数量过多(α=0.05),为每个基因分配一个 p 值。我们还使用 EVA 根据显著基因数量过多(α=0.05),为每个 GO 组分配一个 p 值。如果一个 GO 类别在两个研究中都达到显著水平(α=0.05),则该类别被认为是可复制的。我们发现了两个在两个研究中都可复制的 GO 类别。第一个是“细胞成分组织和生物发生的调节”,是一个 GO 生物学过程,在检测和复制研究中的 p 值分别为 0.010 和 0.014。第二个是“肌动蛋白细胞骨架”,是一个 GO 细胞成分,在检测和复制研究中的 p 值分别为 0.040 和 0.046。
对两项散发性肌萎缩侧索硬化症全基因组关联研究中两两遗传关联的途径分析显示,一组参与细胞成分组织和肌动蛋白细胞骨架的基因,特别是先前的全基因组关联研究未报道的基因。然而,先前的生物学研究已经将肌动蛋白细胞骨架与肌萎缩侧索硬化症和其他运动神经元疾病联系起来。本研究支持这样一种观点,即全基因组关联研究数据的途径水平分析可能会发现一些重要的关联,而这些关联是使用传统的逐个 SNP 方法无法揭示的。