Department of Paediatrics and Adolescent Medicine, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong SAR, China.
PLoS One. 2012;7(7):e41802. doi: 10.1371/journal.pone.0041802. Epub 2012 Jul 25.
Dravet syndrome is a severe form of epilepsy. Majority of patients have a mutation in SCN1A gene, which encodes a voltage-gated sodium channel. A recent study has demonstrated that 16% of SCN1A-negative patients have a mutation in PCDH19, the gene encoding protocadherin-19. Mutations in other genes account for only a very small proportion of families. TSPYL4 is a novel candidate gene within the locus 6q16.3-q22.31 identified by linkage study.
The present study examined the mutations in epileptic Chinese children with emphasis on Dravet syndrome.
A hundred children with severe epilepsy were divided into Dravet syndrome and non-Dravet syndrome groups and screened for SCN1A mutations by direct sequencing. SCN1A-negative Dravet syndrome patients and patients with phenotypes resembling Dravet syndrome were checked for PCDH19 and TSPYL4 mutations.
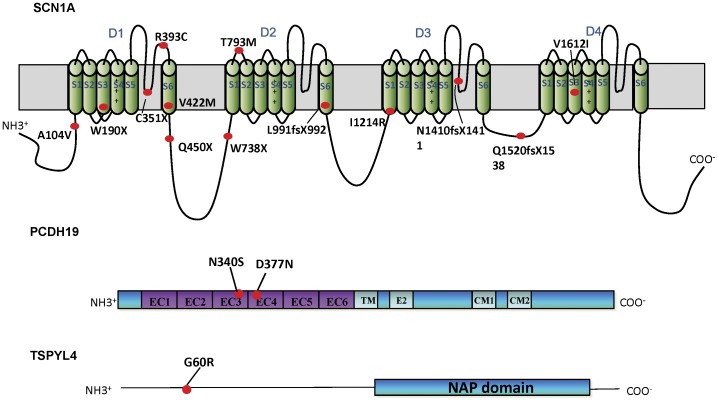
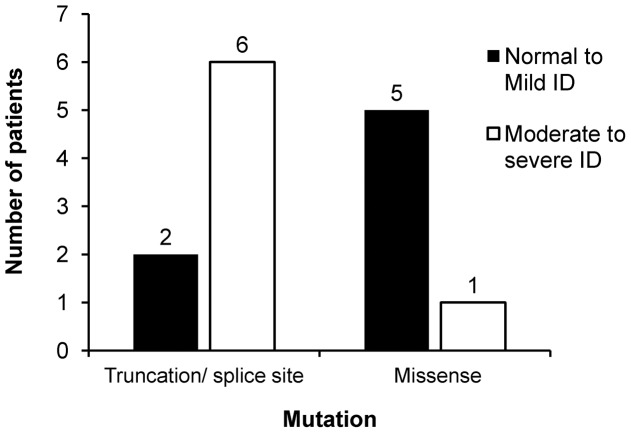
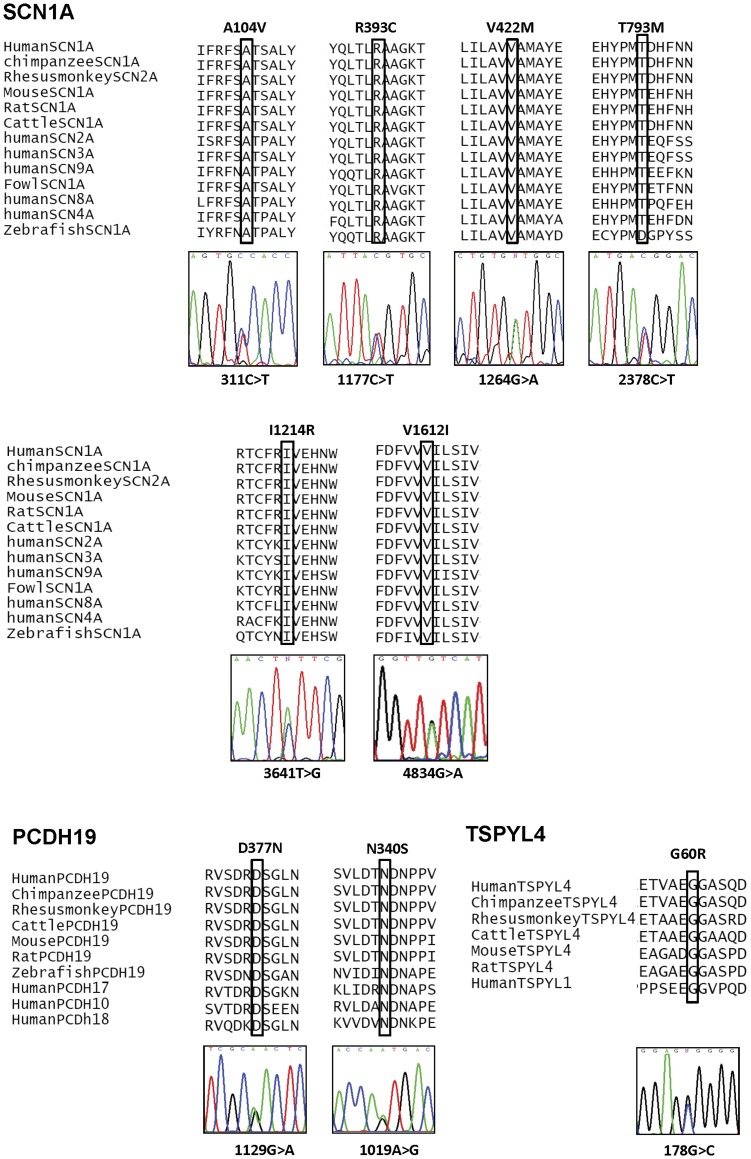
Eighteen patients (9 males, 9 females) were diagnosed to have Dravet syndrome. Among them, 83% (15/18) had SCN1A mutations including truncating (7), splice site (2) and missense mutations (6). The truncating/splice site mutations were associated with moderate to severe degree of intellectual disability (p<0.05). During the progression of disease, 73% (11/15) had features fitting into the diagnostic criteria of autism spectrum disorder and 53% (8/15) had history of vaccination-induced seizures. A novel PCDH19 p.D377N mutation was identified in one SCN1A-negative female patient with Dravet syndrome and a known PCDH19 p.N340S mutation in a female non-Dravet syndrome patient. The former also inherited a TSPYL4 p.G60R variant.
A high percentage of SCN1A mutations was identified in our Chinese cohort of Dravet syndrome patients but none in the rest of patients. We demonstrated that truncating/splice site mutations were linked to moderate to severe intellectual disability in these patients. A de novo PCDH19 missense mutation together with an inherited TSPYL4 missense variant were identified in a patient with Dravet syndrome.
Dravet 综合征是一种严重的癫痫形式。大多数患者的 SCN1A 基因发生突变,该基因编码电压门控钠离子通道。最近的一项研究表明,16%的 SCN1A 阴性患者的 PCDH19 基因突变,PCDH19 基因编码原钙黏蛋白-19。其他基因突变仅占极少数家庭。TSPYL4 是通过连锁研究在 6q16.3-q22.31 区域鉴定的一个新的候选基因。
本研究重点研究了患有癫痫的中国儿童的突变情况,特别是 Dravet 综合征。
将 100 名患有严重癫痫的儿童分为 Dravet 综合征组和非 Dravet 综合征组,并通过直接测序筛查 SCN1A 突变。对 SCN1A 阴性 Dravet 综合征患者和表型类似 Dravet 综合征的患者进行 PCDH19 和 TSPYL4 突变检测。
18 名患者(9 名男性,9 名女性)被诊断为 Dravet 综合征。其中,83%(15/18)的患者存在 SCN1A 突变,包括截断(7 个)、剪接位点(2 个)和错义突变(6 个)。截断/剪接位点突变与中重度智力残疾相关(p<0.05)。在疾病进展过程中,73%(11/15)具有符合自闭症谱系障碍诊断标准的特征,53%(8/15)有疫苗诱导癫痫发作的病史。在一名 SCN1A 阴性的 Dravet 综合征女性患者中发现了一种新的 PCDH19 p.D377N 突变,在一名非 Dravet 综合征女性患者中发现了一种已知的 PCDH19 p.N340S 突变。前者还遗传了 TSPYL4 p.G60R 变异。
在我们的中国 Dravet 综合征患者队列中发现了很高比例的 SCN1A 突变,但在其他患者中没有发现。我们证明,这些患者的截断/剪接位点突变与中重度智力残疾相关。在一名 Dravet 综合征患者中发现了一种新的 PCDH19 错义突变,同时还遗传了一种 TSPYL4 错义变异。