Curtis David
Centre for Psychiatry, Barts and the London School of Medicine and Dentistry, London, UK.
Adv Appl Bioinform Chem. 2012;5:1-9. doi: 10.2147/AABC.S33049. Epub 2012 Jul 24.
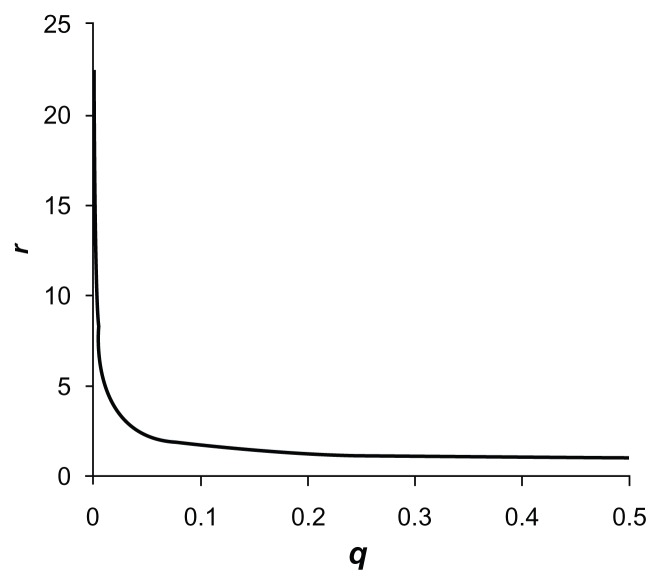
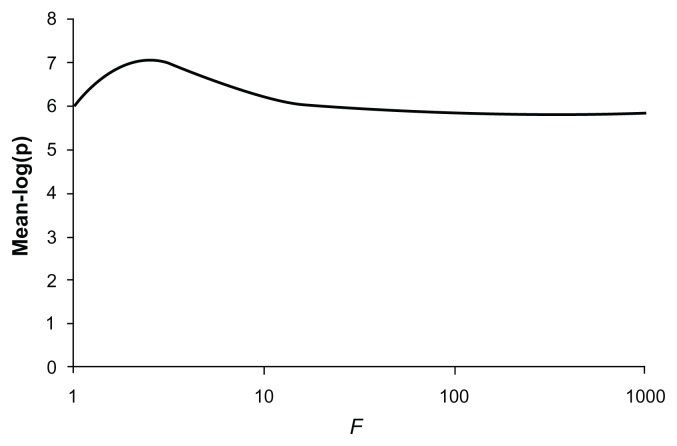
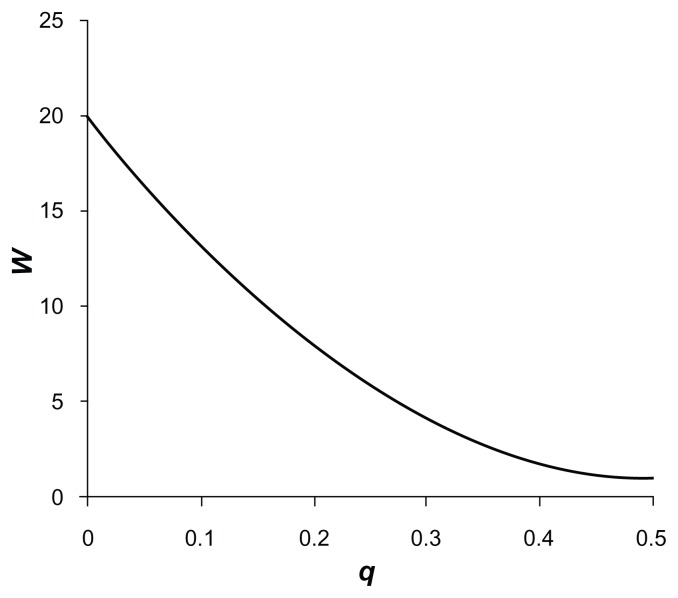
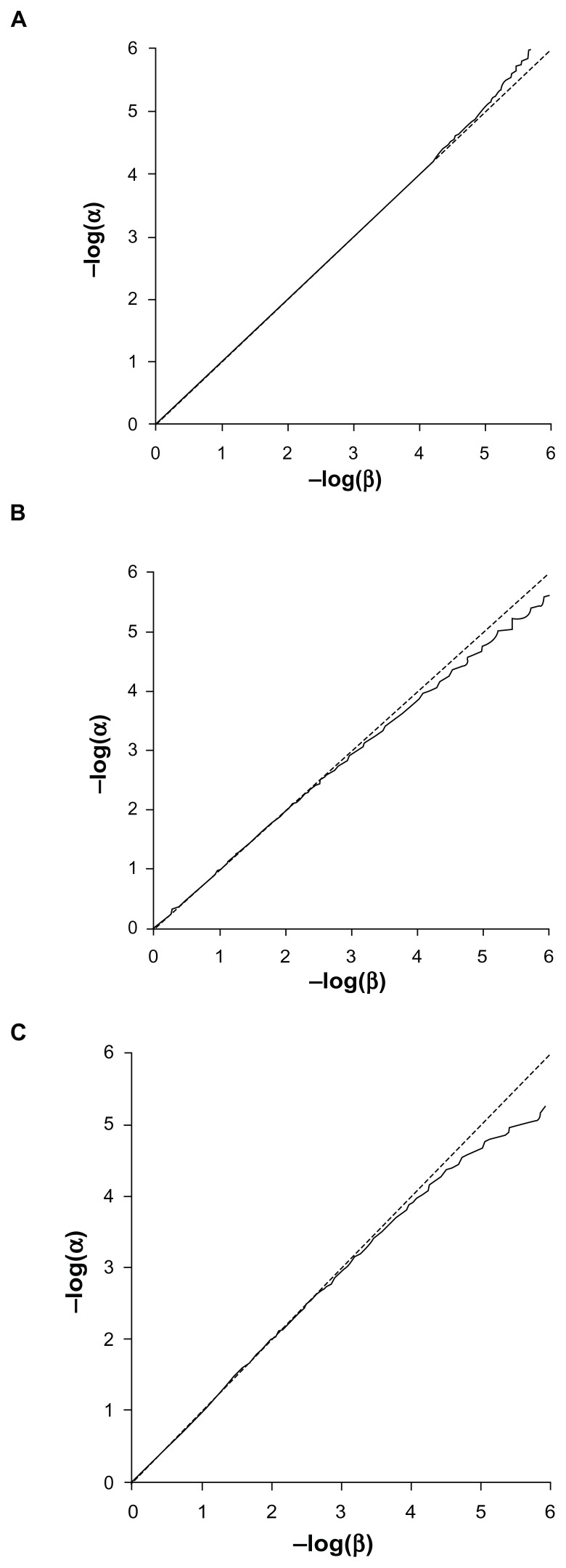
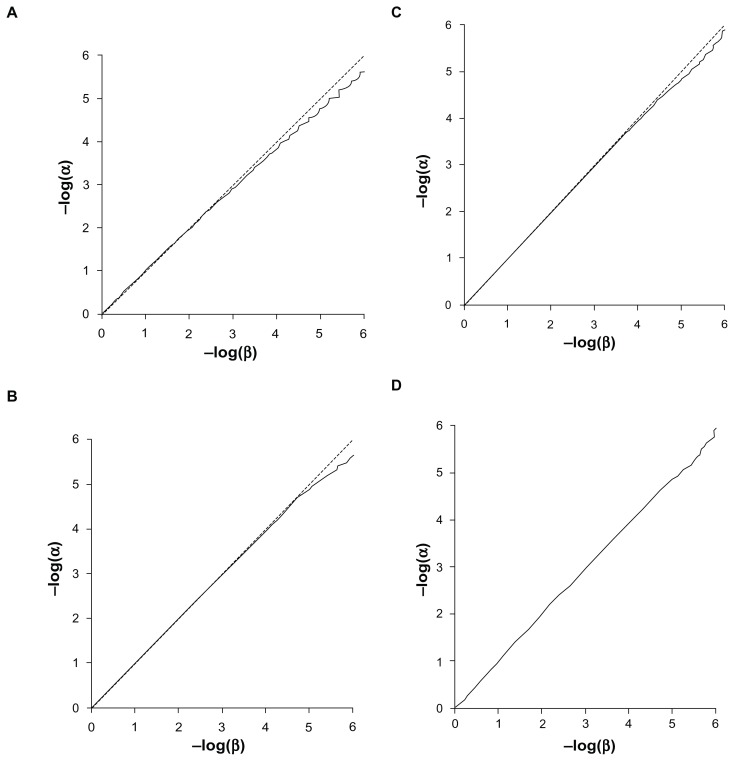
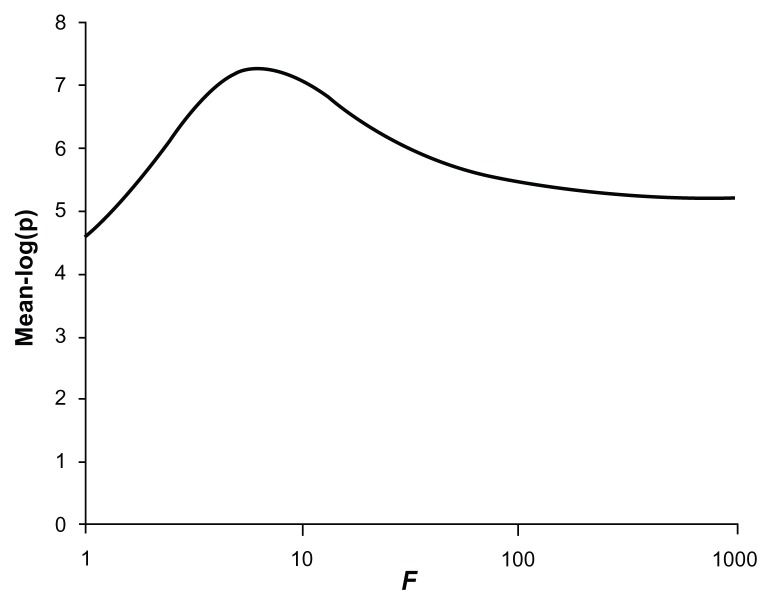
Previously described methods for the combined analysis of common and rare variants have disadvantages such as requiring an arbitrary classification of variants or permutation testing to assess statistical significance. Here we propose a novel method which implements a weighting scheme based on allele frequencies observed in both cases and controls. Because the test is unbiased, scores can be analyzed with a standard t-test. To test its validity we applied it to data for common, rare, and very rare variants simulated under the null hypothesis. To test its power we applied it to simulated data in which association was present, including data using the observed allele frequencies of common and rare variants in NOD2 previously reported in cases of Crohn's disease and controls. The method produced results that conformed well to those expected under the null hypothesis. It demonstrated more power to detect association when rare and common variants were analyzed jointly, the power further increasing when rare variants were assigned higher weights. 20,000 analyses of a gene containing 62 variants could be performed in 80 minutes on a laptop. This approach shows promise for the analysis of data currently emerging from genome wide sequencing studies.
先前描述的用于常见和罕见变异联合分析的方法存在一些缺点,比如需要对变异进行任意分类或进行置换检验来评估统计显著性。在此,我们提出一种新方法,该方法基于在病例组和对照组中观察到的等位基因频率实施一种加权方案。由于该检验是无偏的,因此可以使用标准t检验来分析分数。为了检验其有效性,我们将其应用于在零假设下模拟的常见、罕见和非常罕见变异的数据。为了检验其效能,我们将其应用于存在关联的模拟数据,包括使用先前在克罗恩病病例和对照中报道的NOD2常见和罕见变异的观察等位基因频率的数据。该方法产生的结果与零假设下预期的结果非常吻合。当联合分析罕见和常见变异时,它显示出更强的检测关联的能力,当为罕见变异赋予更高权重时,效能进一步提高。在一台笔记本电脑上,80分钟内可以对包含62个变异的一个基因进行20,000次分析。这种方法对于分析目前来自全基因组测序研究的数据显示出前景。