Harvard Medical School, Boston, Massachusetts, United States of America.
PLoS Med. 2012;9(7):e1001276. doi: 10.1371/journal.pmed.1001276. Epub 2012 Jul 31.
Policymakers and regulators in the United States (US) and the European Union (EU) are weighing reforms to their medical device approval and post-market surveillance systems. Data may be available that identify strengths and weakness of the approaches to medical device regulation in these settings.
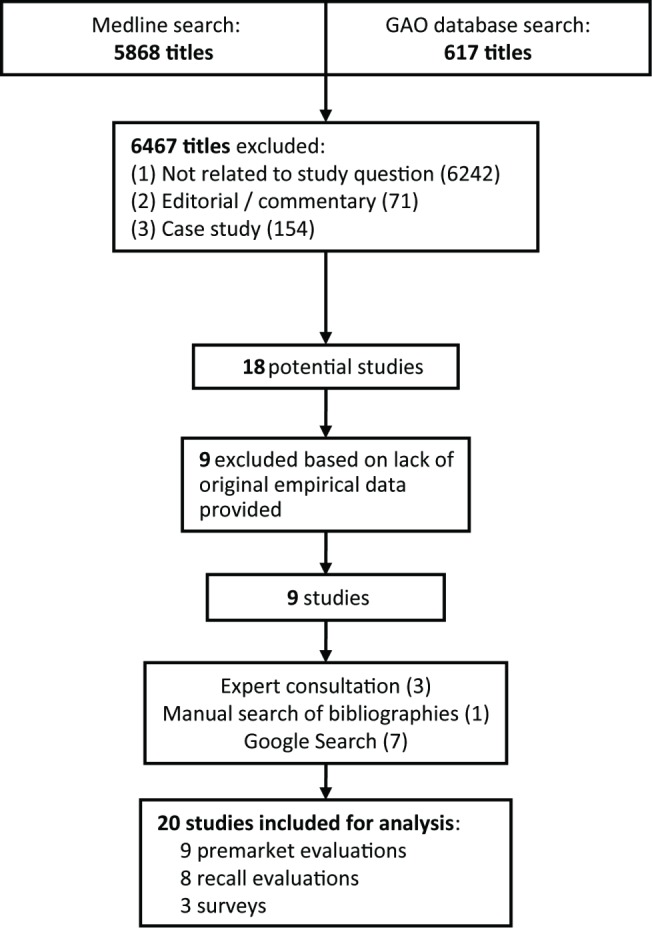
We performed a systematic review to find empirical studies evaluating medical device regulation in the US or EU. We searched Medline using two nested categories that included medical devices and glossary terms attributable to the US Food and Drug Administration and the EU, following PRISMA guidelines for systematic reviews. We supplemented this search with a review of the US Government Accountability Office online database for reports on US Food and Drug Administration device regulation, consultations with local experts in the field, manual reference mining of selected articles, and Google searches using the same key terms used in the Medline search. We found studies of premarket evaluation and timing (n = 9), studies of device recalls (n = 8), and surveys of device manufacturers (n = 3). These studies provide evidence of quality problems in pre-market submissions in the US, provide conflicting views of device safety based largely on recall data, and relay perceptions of some industry leaders from self-surveys.
Few studies have quantitatively assessed medical device regulation in either the US or EU. Existing studies of US and EU device approval and post-market evaluation performance suggest that policy reforms are necessary for both systems, including improving classification of devices in the US and promoting transparency and post-market oversight in the EU. Assessment of regulatory performance in both settings is limited by lack of data on post-approval safety outcomes. Changes to these device approval and post-marketing systems must be accompanied by ongoing research to ensure that there is better assessment of what works in either setting.
美国(US)和欧盟(EU)的政策制定者和监管机构正在权衡对其医疗器械审批和上市后监测系统进行改革。可能有数据可以确定这两个地区医疗器械监管方法的优势和劣势。
我们进行了一项系统评价,以寻找评估美国或欧盟医疗器械监管的实证研究。我们按照 PRISMA 系统评价指南,使用包含医疗器械和可归因于美国食品和药物管理局和欧盟的术语表的两个嵌套类别,在 Medline 上进行了搜索。我们通过查阅美国政府问责局在线数据库中有关美国食品和药物管理局设备监管的报告、与该领域当地专家进行咨询、对选定文章进行手动参考挖掘以及使用与 Medline 搜索相同的关键词进行谷歌搜索,补充了这项搜索。我们找到了 9 项关于上市前评估和时间的研究、8 项关于设备召回的研究和 3 项关于设备制造商的调查。这些研究提供了美国上市前提交材料中存在质量问题的证据,根据召回数据提供了对设备安全性的相互矛盾的看法,并从自我调查中反映了一些行业领导者的看法。
很少有研究对美国或欧盟的医疗器械监管进行定量评估。现有的关于美国和欧盟设备审批和上市后评估绩效的研究表明,两个系统都需要政策改革,包括改进美国设备的分类和促进欧盟的透明度和上市后监督。这两个地区监管绩效的评估都受到缺乏批准后安全结果数据的限制。对这些设备审批和上市后监管系统的改变必须伴随着持续的研究,以确保对任何一个地区有效的方法进行更好的评估。