Quantitative Sciences, GlaxoSmithKline, Research Triangle Park, North Carolina, United States of America.
PLoS One. 2012;7(8):e42530. doi: 10.1371/journal.pone.0042530. Epub 2012 Aug 9.
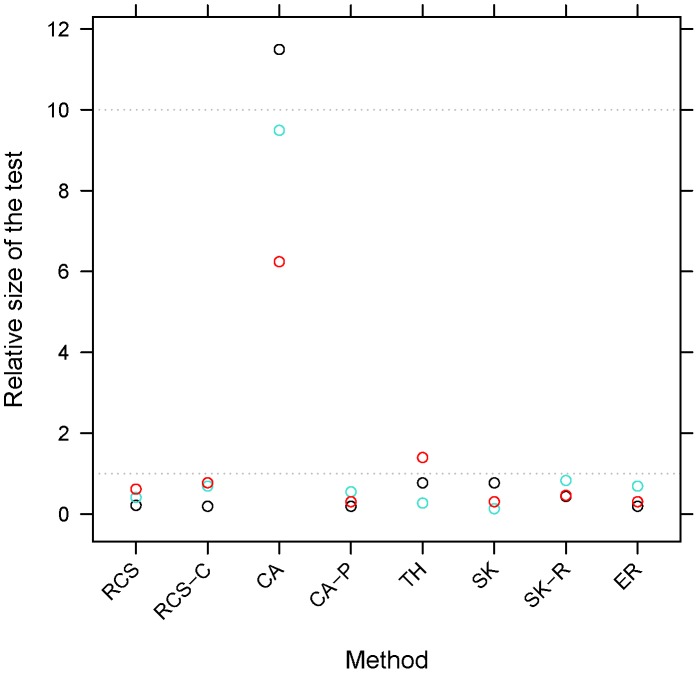
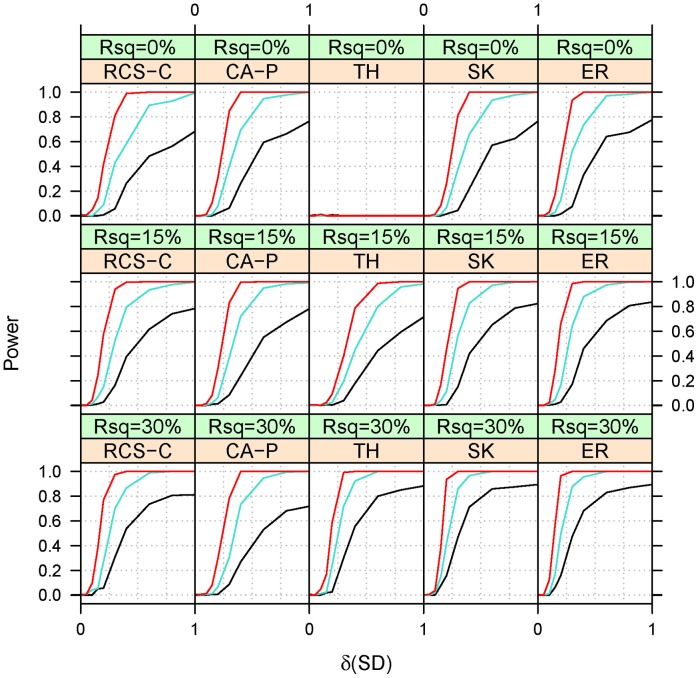
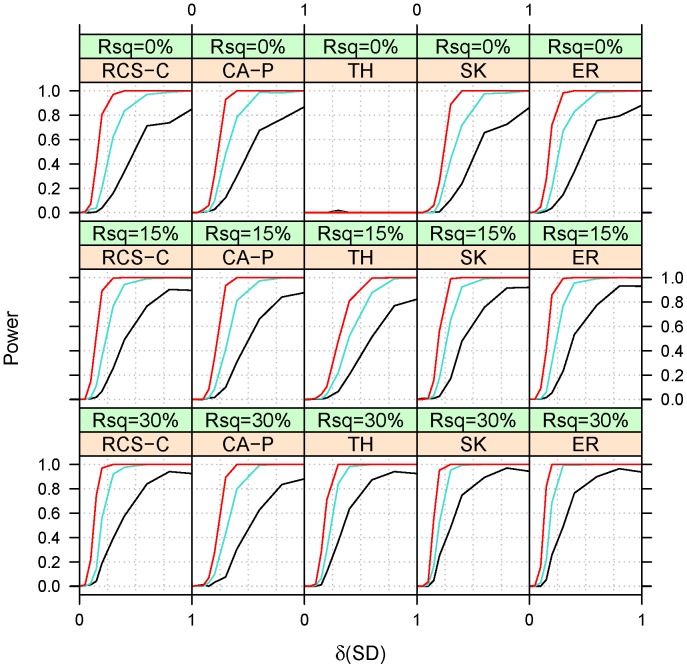
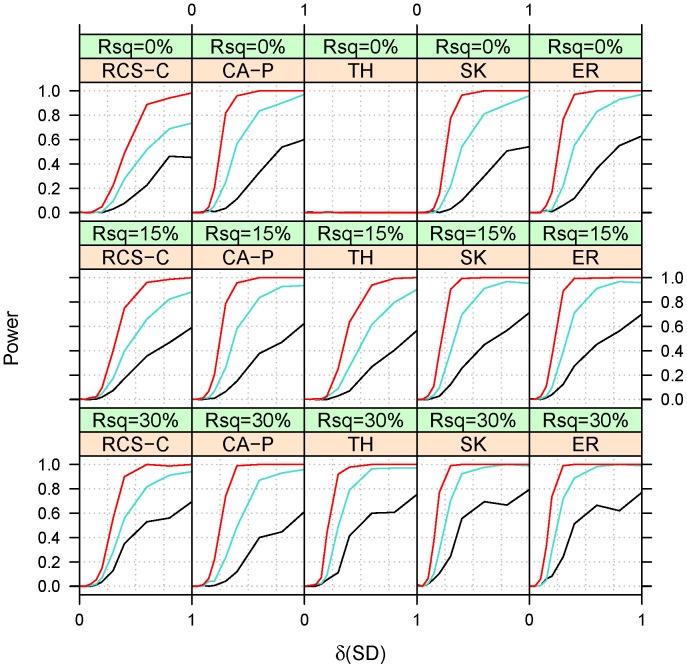
Genome-wide association studies have found thousands of common genetic variants associated with a wide variety of diseases and other complex traits. However, a large portion of the predicted genetic contribution to many traits remains unknown. One plausible explanation is that some of the missing variation is due to the effects of rare variants. Nonetheless, the statistical analysis of rare variants is challenging. A commonly used method is to contrast, within the same region (gene), the frequency of minor alleles at rare variants between cases and controls. However, this strategy is most useful under the assumption that the tested variants have similar effects. We previously proposed a method that can accommodate heterogeneous effects in the analysis of quantitative traits. Here we extend this method to include binary traits that can accommodate covariates. We use simulations for a variety of causal and covariate impact scenarios to compare the performance of the proposed method to standard logistic regression, C-alpha, SKAT, and EREC. We found that i) logistic regression methods perform well when the heterogeneity of the effects is not extreme and ii) SKAT and EREC have good performance under all tested scenarios but they can be computationally intensive. Consequently, it would be more computationally desirable to use a two-step strategy by (i) selecting promising genes by faster methods and ii) analyzing selected genes using SKAT/EREC. To select promising genes one can use (1) regression methods when effect heterogeneity is assumed to be low and the covariates explain a non-negligible part of trait variability, (2) C-alpha when heterogeneity is assumed to be large and covariates explain a small fraction of trait's variability and (3) the proposed trend and heterogeneity test when the heterogeneity is assumed to be non-trivial and the covariates explain a large fraction of trait variability.
全基因组关联研究发现了数千种与多种疾病和其他复杂特征相关的常见遗传变异。然而,许多特征的预测遗传贡献的很大一部分仍然未知。一个合理的解释是,一些缺失的变异是由于罕见变异的影响。尽管如此,罕见变异的统计分析仍然具有挑战性。一种常用的方法是在同一区域(基因)内,比较病例和对照之间罕见变异的次要等位基因频率。然而,这种策略在测试变体具有相似影响的假设下最有用。我们之前提出了一种可以在分析定量特征时容纳异质性效应的方法。在这里,我们将该方法扩展到包括可以容纳协变量的二元特征。我们使用各种因果和协变量影响场景的模拟来比较所提出的方法与标准逻辑回归、C-alpha、SKAT 和 EREC 的性能。我们发现:i)当效应异质性不是极端时,逻辑回归方法的性能良好;ii)SKAT 和 EREC 在所有测试场景下都具有良好的性能,但它们可能计算密集。因此,使用两步策略更为理想,即通过 i)使用更快的方法选择有前途的基因,ii)使用 SKAT/EREC 分析选定的基因。要选择有前途的基因,可以使用 1)当假设效应异质性较低且协变量解释了特征可变性的不可忽略部分时使用回归方法,2)当假设异质性较大且协变量解释了特征可变性的一小部分时使用 C-alpha,以及 3)当假设异质性不可忽视且协变量解释了特征可变性的很大一部分时使用拟议的趋势和异质性检验。