Lee Selyeong, Won Sungho, Kim Young Jin, Kim Yongkang, Kim Bong-Jo, Park Taesung
Department of Statistics, Seoul National University, Seoul, Korea.
Graduate School of Public Health, Seoul National University, Seoul, Korea.
Genet Epidemiol. 2017 Apr;41(3):198-209. doi: 10.1002/gepi.22021. Epub 2016 Dec 31.
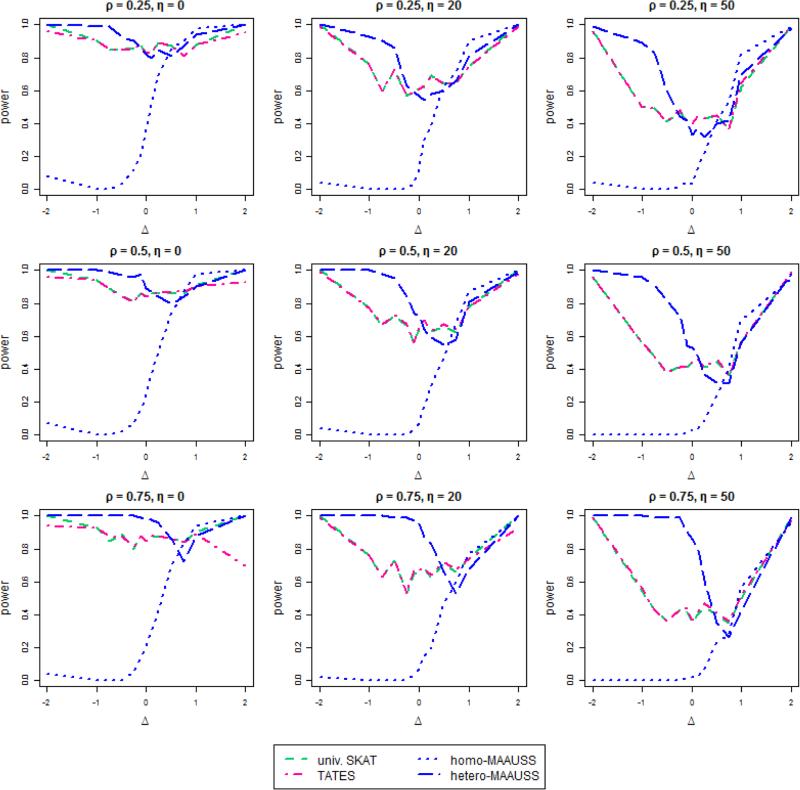
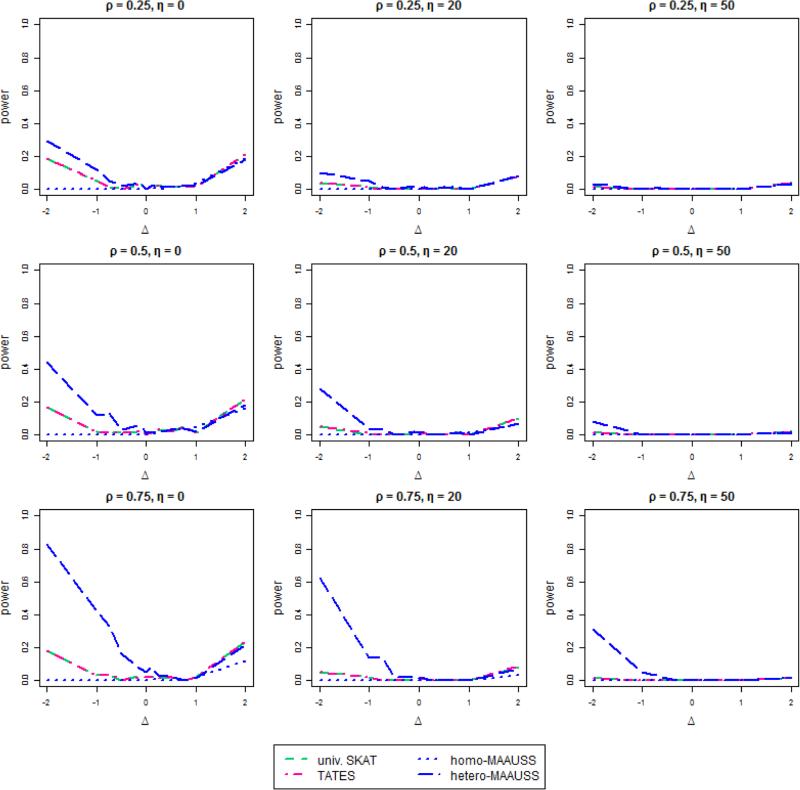
Although genome-wide association studies (GWAS) have now discovered thousands of genetic variants associated with common traits, such variants cannot explain the large degree of "missing heritability," likely due to rare variants. The advent of next generation sequencing technology has allowed rare variant detection and association with common traits, often by investigating specific genomic regions for rare variant effects on a trait. Although multiple correlated phenotypes are often concurrently observed in GWAS, most studies analyze only single phenotypes, which may lessen statistical power. To increase power, multivariate analyses, which consider correlations between multiple phenotypes, can be used. However, few existing multivariant analyses can identify rare variants for assessing multiple phenotypes. Here, we propose Multivariate Association Analysis using Score Statistics (MAAUSS), to identify rare variants associated with multiple phenotypes, based on the widely used sequence kernel association test (SKAT) for a single phenotype. We applied MAAUSS to whole exome sequencing (WES) data from a Korean population of 1,058 subjects to discover genes associated with multiple traits of liver function. We then assessed validation of those genes by a replication study, using an independent dataset of 3,445 individuals. Notably, we detected the gene ZNF620 among five significant genes. We then performed a simulation study to compare MAAUSS's performance with existing methods. Overall, MAAUSS successfully conserved type 1 error rates and in many cases had a higher power than the existing methods. This study illustrates a feasible and straightforward approach for identifying rare variants correlated with multiple phenotypes, with likely relevance to missing heritability.
尽管全基因组关联研究(GWAS)现已发现数千种与常见性状相关的基因变异,但此类变异无法解释很大程度的“缺失遗传力”,这可能是由罕见变异导致的。下一代测序技术的出现使得罕见变异得以检测,并与常见性状相关联,通常是通过研究特定基因组区域中罕见变异对某一性状的影响。尽管在GWAS中经常同时观察到多种相关表型,但大多数研究仅分析单一表型,这可能会降低统计效力。为了提高效力,可以使用考虑多种表型之间相关性的多变量分析。然而,现有的多变量分析很少能识别出用于评估多种表型的罕见变异。在此,我们提出基于单表型广泛使用的序列核关联检验(SKAT)的多变量评分统计关联分析(MAAUSS),以识别与多种表型相关的罕见变异。我们将MAAUSS应用于来自1058名韩国受试者的全外显子组测序(WES)数据,以发现与肝功能多种性状相关的基因。然后,我们使用一个3445人的独立数据集,通过重复研究评估这些基因的验证情况。值得注意的是,我们在五个显著基因中检测到了ZNF620基因。然后,我们进行了一项模拟研究,以比较MAAUSS与现有方法的性能。总体而言,MAAUSS成功保持了I型错误率,并且在许多情况下比现有方法具有更高的效力。这项研究说明了一种可行且直接的方法,用于识别与多种表型相关的罕见变异,这可能与缺失遗传力相关。