Astbury Centre for Structural Molecular Biology, University of Leeds, Leeds, United Kingdom.
PLoS One. 2012;7(8):e43253. doi: 10.1371/journal.pone.0043253. Epub 2012 Aug 20.
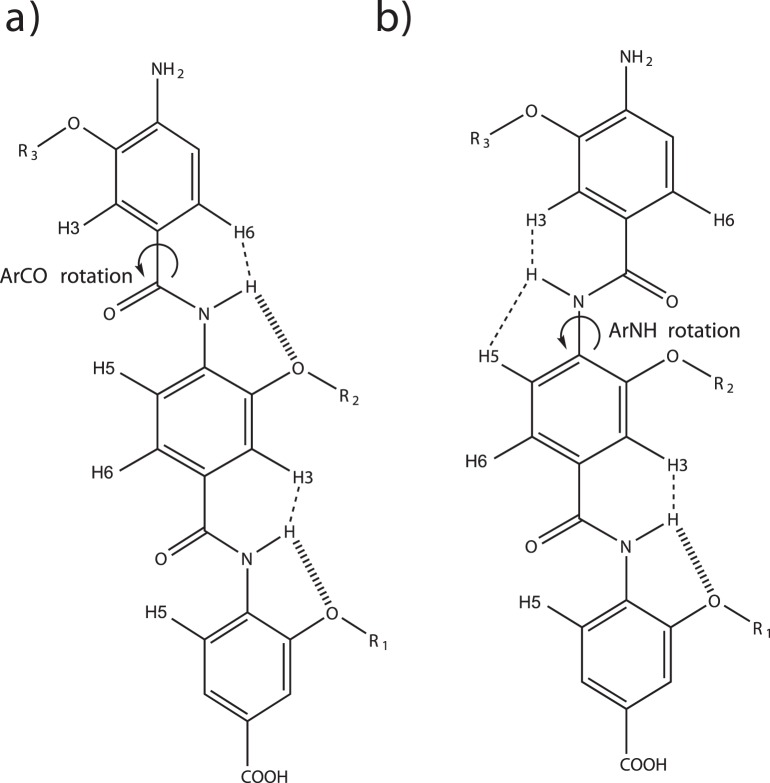
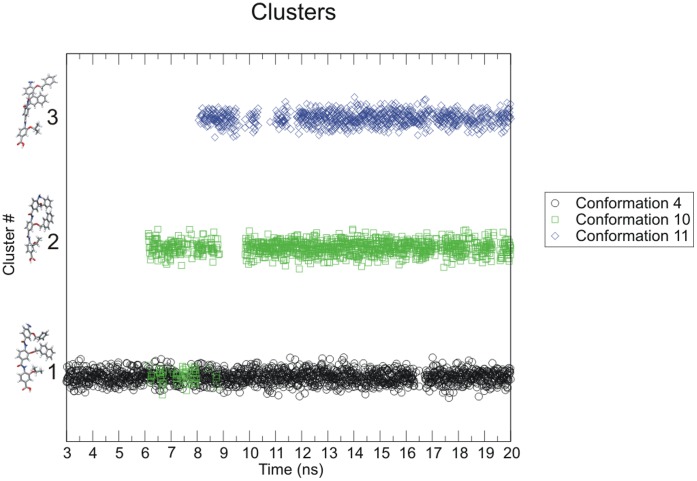
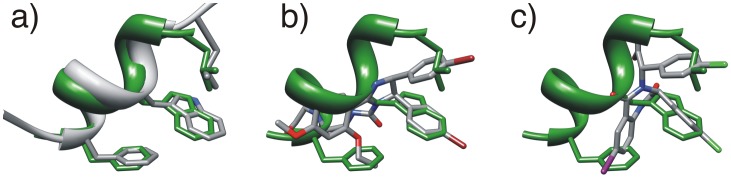
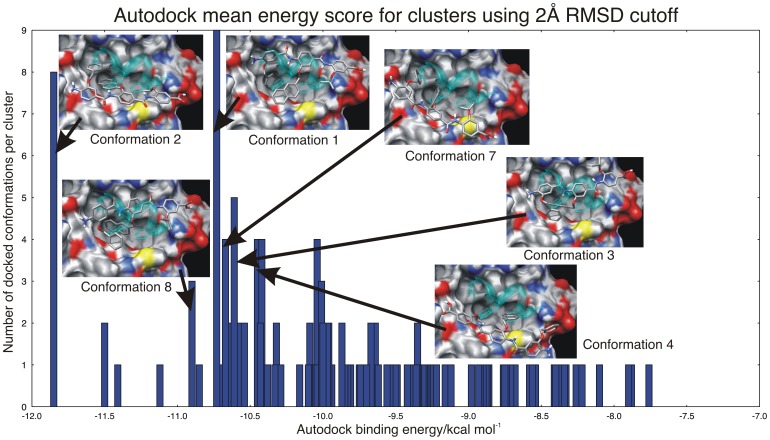
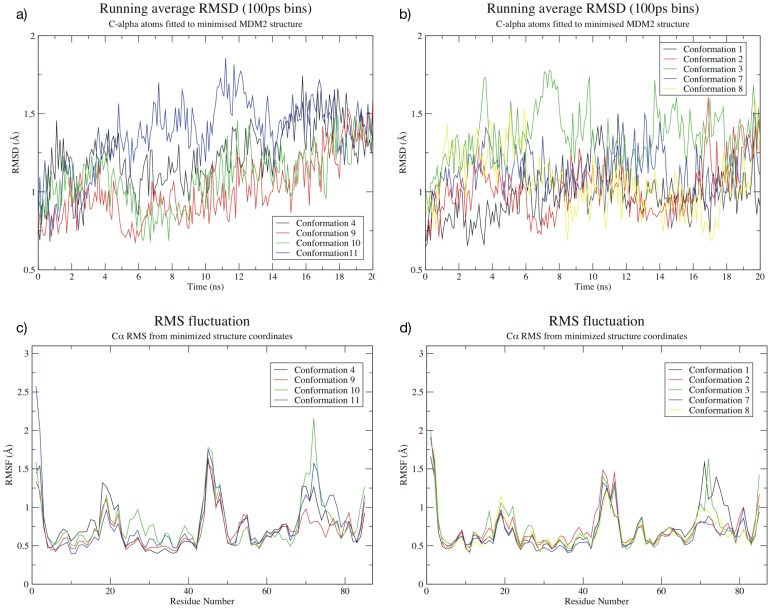
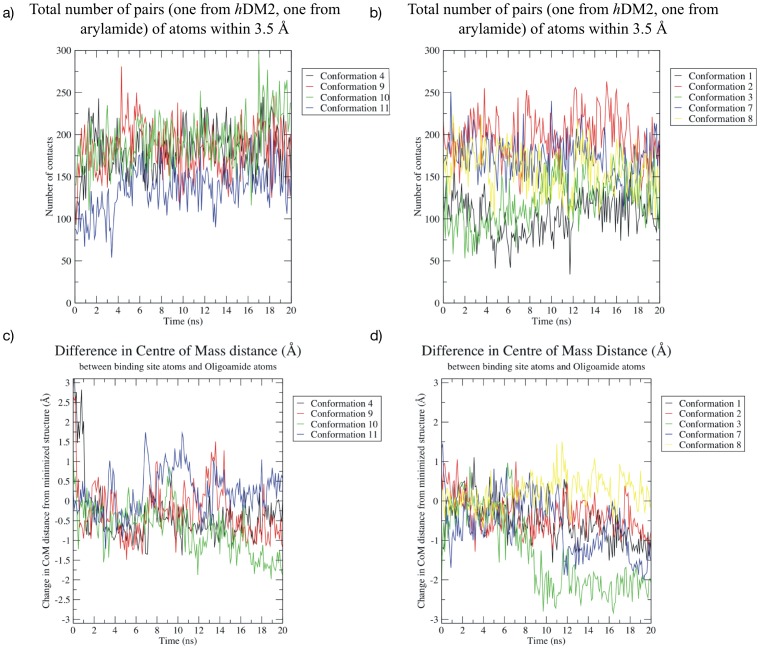
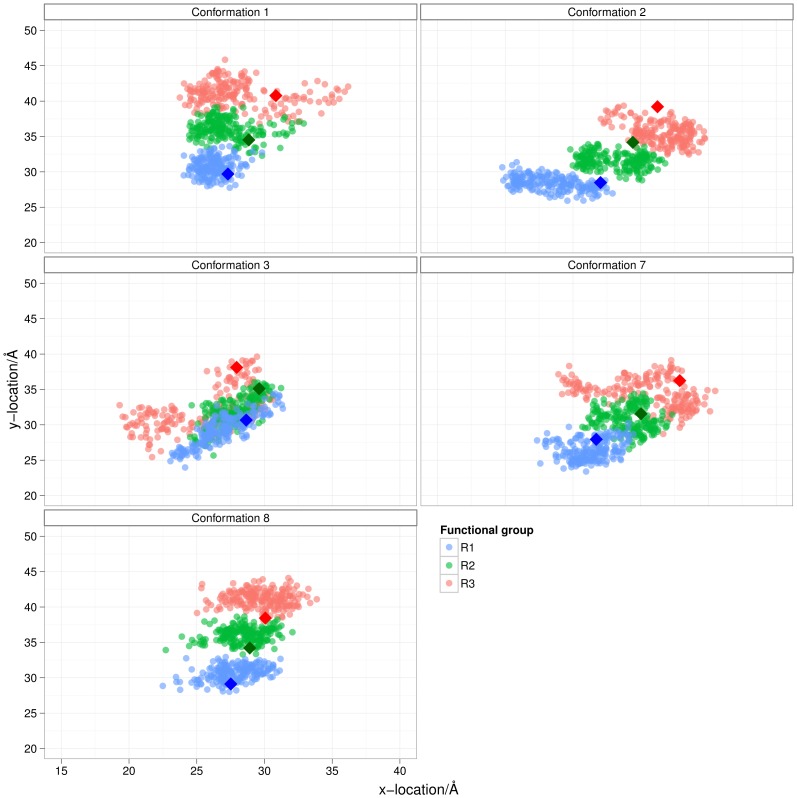
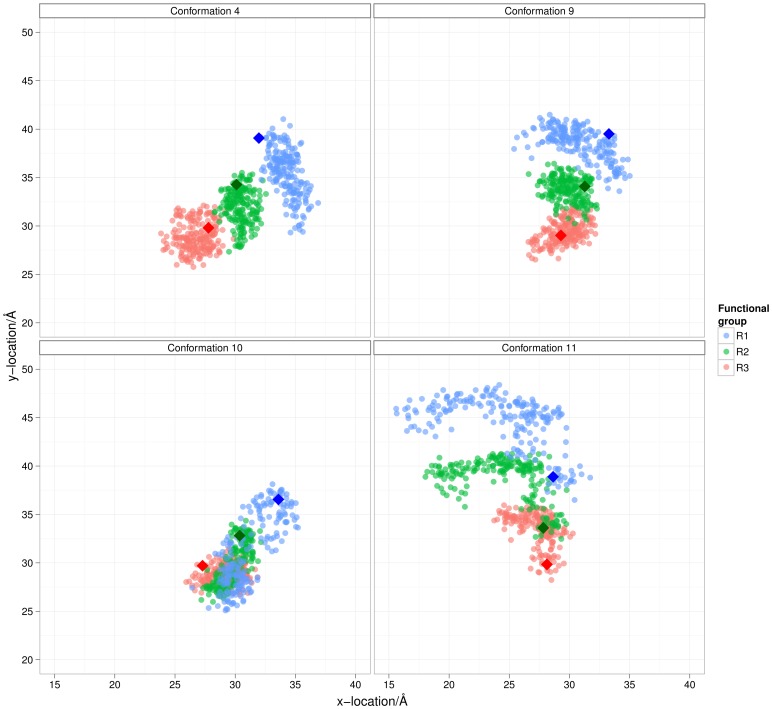
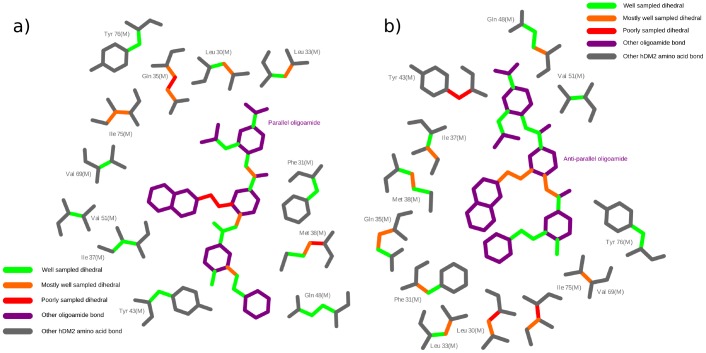
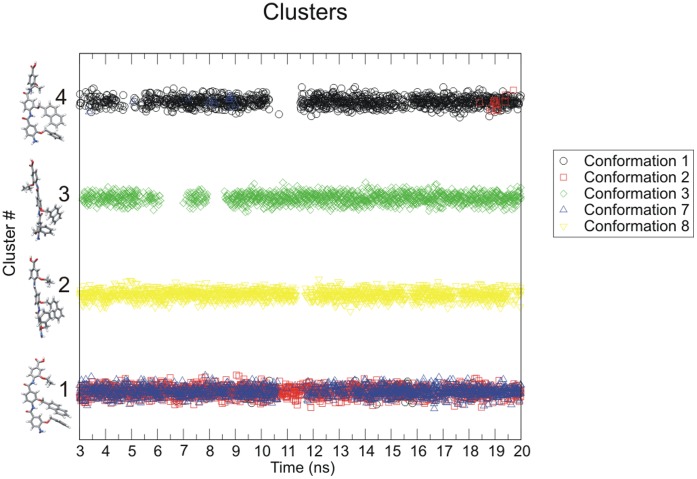
The design of novel α-helix mimetic inhibitors of protein-protein interactions is of interest to pharmaceuticals and chemical genetics researchers as these inhibitors provide a chemical scaffold presenting side chains in the same geometry as an α-helix. This conformational arrangement allows the design of high affinity inhibitors mimicking known peptide sequences binding specific protein substrates. We show that GAFF and AutoDock potentials do not properly capture the conformational preferences of α-helix mimetics based on arylamide oligomers and identify alternate parameters matching solution NMR data and suitable for molecular dynamics simulation of arylamide compounds. Results from both docking and molecular dynamics simulations are consistent with the arylamides binding in the p53 peptide binding pocket. Simulations of arylamides in the p53 binding pocket of hDM2 are consistent with binding, exhibiting similar structural dynamics in the pocket as simulations of known hDM2 binders Nutlin-2 and a benzodiazepinedione compound. Arylamide conformations converge towards the same region of the binding pocket on the 20 ns time scale, and most, though not all dihedrals in the binding pocket are well sampled on this timescale. We show that there are two putative classes of binding modes for arylamide compounds supported equally by the modeling evidence. In the first, the arylamide compound lies parallel to the observed p53 helix. In the second class, not previously identified or proposed, the arylamide compound lies anti-parallel to the p53 helix.
新型α-螺旋模拟蛋白-蛋白相互作用抑制剂的设计引起了制药和化学遗传学研究人员的兴趣,因为这些抑制剂提供了一个化学支架,其侧链呈现出与α-螺旋相同的几何形状。这种构象排列允许设计具有高亲和力的抑制剂,模拟已知的肽序列与特定的蛋白质底物结合。我们表明,GAFF 和 AutoDock 势能不能正确捕捉基于芳酰胺低聚物的α-螺旋模拟物的构象偏好,并确定了与溶液 NMR 数据匹配的替代参数,适合芳酰胺化合物的分子动力学模拟。对接和分子动力学模拟的结果都与芳酰胺在 p53 肽结合口袋中的结合一致。芳酰胺在 hDM2 的 p53 结合口袋中的模拟与结合一致,在口袋中的结构动力学与已知的 hDM2 结合物 Nutlin-2 和苯并二氮杂酮化合物的模拟相似。芳酰胺构象在 20 ns 的时间尺度上收敛到结合口袋的同一区域,并且在该时间尺度上,虽然不是所有的二面角都在结合口袋中得到很好的采样。我们表明,有两种假定的芳酰胺化合物结合模式,这两种模式都得到了建模证据的支持。在第一种模式中,芳酰胺化合物与观察到的 p53 螺旋平行。在第二种以前未被识别或提出的模式中,芳酰胺化合物与 p53 螺旋反平行。