Department of Computer Science, University of Missouri Columbia, MO, USA.
Front Plant Sci. 2012 Aug 21;3:186. doi: 10.3389/fpls.2012.00186. eCollection 2012.
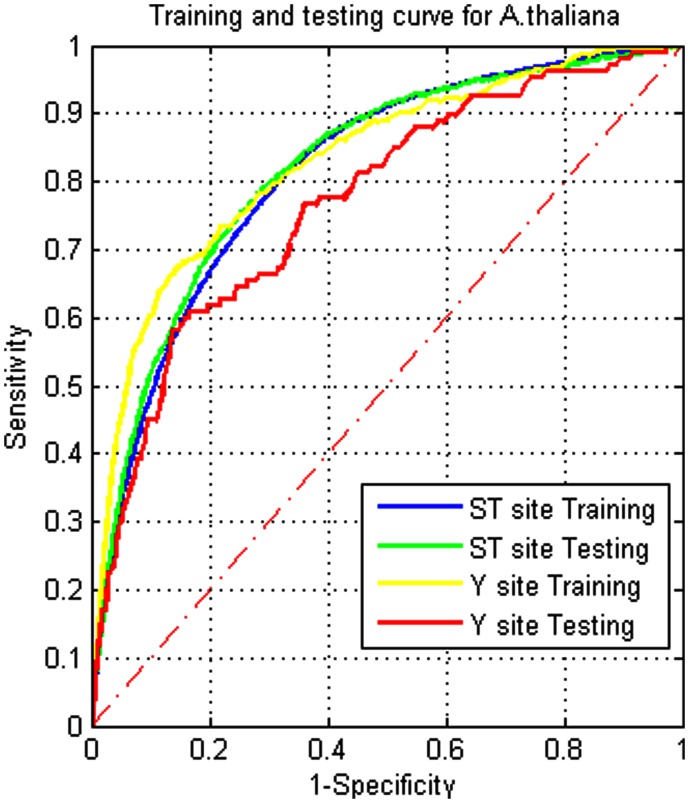
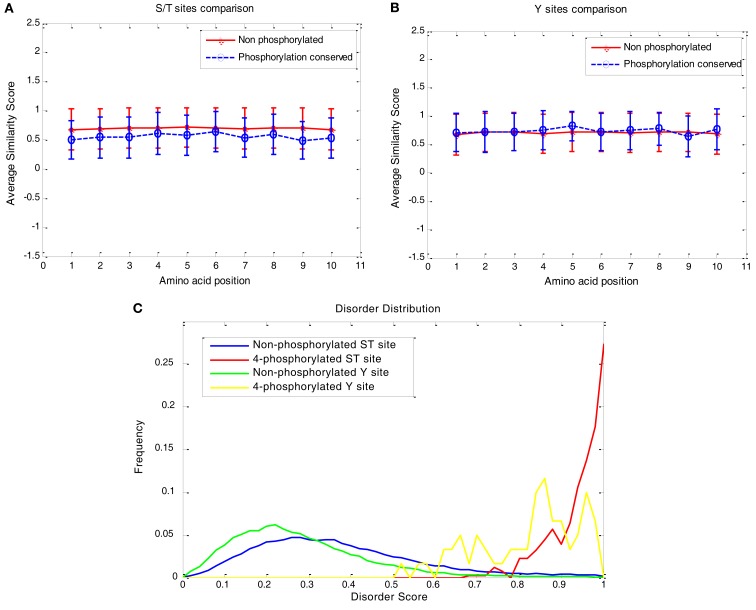
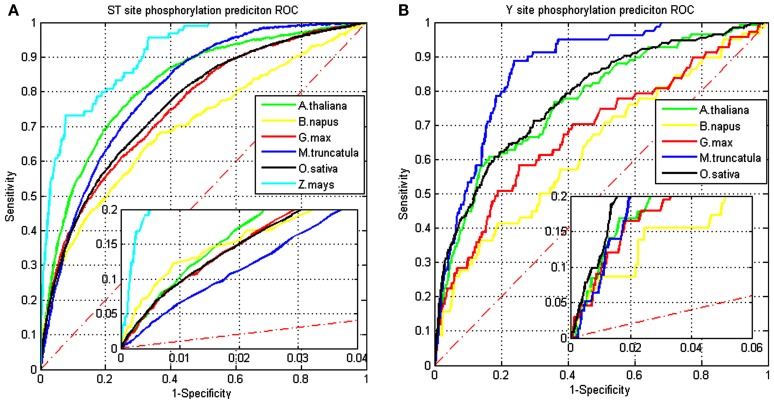
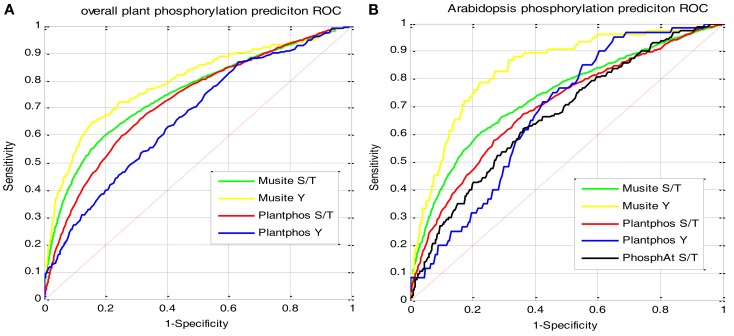
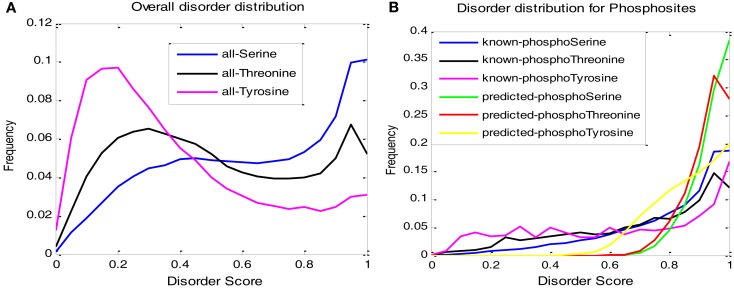
Although protein phosphorylation sites can be reliably identified with high-resolution mass spectrometry, the experimental approach is time-consuming and resource-dependent. Furthermore, it is unlikely that an experimental approach could catalog an entire phosphoproteome. Computational prediction of phosphorylation sites provides an efficient and flexible way to reveal potential phosphorylation sites and provide hypotheses in experimental design. Musite is a tool that we previously developed to predict phosphorylation sites based solely on protein sequence. However, it was not comprehensively applied to plants. In this study, the phosphorylation data from Arabidopsis thaliana, B. napus, G. max, M. truncatula, O. sativa, and Z. mays were collected for cross-species testing and the overall plant-specific prediction as well. The results show that the model for A. thaliana can be extended to other organisms, and the overall plant model from Musite outperforms the current plant-specific prediction tools, Plantphos, and PhosphAt, in prediction accuracy. Furthermore, a comparative study of predicted phosphorylation sites across orthologs among different plants was conducted to reveal potential evolutionary features. A bipolar distribution of isolated, non-conserved phosphorylation sites, and highly conserved ones in terms of the amino acid type was observed. It also shows that predicted phosphorylation sites conserved within orthologs do not necessarily share more sequence similarity in the flanking regions than the background, but they often inherit protein disorder, a property that does not necessitate high sequence conservation. Our analysis also suggests that the phosphorylation frequencies among serine, threonine, and tyrosine correlate with their relative proportion in disordered regions. Musite can be used as a web server (http://musite.net) or downloaded as an open-source standalone tool (http://musite.sourceforge.net/).
虽然蛋白质磷酸化位点可以通过高分辨率质谱可靠地鉴定,但实验方法耗时且依赖资源。此外,实验方法不太可能对整个磷酸化组进行编目。磷酸化位点的计算预测提供了一种高效灵活的方法来揭示潜在的磷酸化位点,并为实验设计提供假设。Musite 是我们之前开发的一种仅基于蛋白质序列预测磷酸化位点的工具。然而,它并没有被全面应用于植物。在这项研究中,我们收集了拟南芥、油菜、大豆、蒺藜苜蓿、水稻和玉米的磷酸化数据,用于交叉物种测试和整体植物特异性预测。结果表明,拟南芥模型可以扩展到其他生物体,并且 Musite 的整体植物模型在预测准确性方面优于当前的植物特异性预测工具 Plantphos 和 PhosphAt。此外,还对不同植物中同源物的预测磷酸化位点进行了比较研究,以揭示潜在的进化特征。观察到孤立的、非保守的磷酸化位点和氨基酸类型高度保守的两极分布。这也表明,在同源物中保守的预测磷酸化位点在侧翼区域不一定比背景具有更多的序列相似性,但它们通常继承蛋白质无序性,这种特性不需要高度的序列保守性。我们的分析还表明,丝氨酸、苏氨酸和酪氨酸的磷酸化频率与其在无序区域中的相对比例相关。Musite 可以用作网络服务器(http://musite.net)或下载为开源独立工具(http://musite.sourceforge.net/)。