Department of Computer Science and Engineering, Yuan Ze University, Chungli 320, Taiwan.
BMC Bioinformatics. 2011 Jun 26;12:261. doi: 10.1186/1471-2105-12-261.
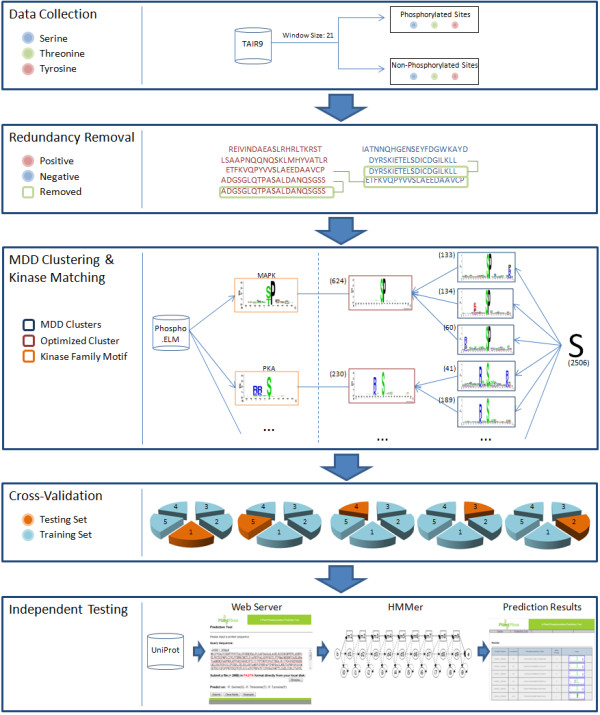
Protein phosphorylation catalyzed by kinases plays crucial regulatory roles in intracellular signal transduction. Due to the difficulty in performing high-throughput mass spectrometry-based experiment, there is a desire to predict phosphorylation sites using computational methods. However, previous studies regarding in silico prediction of plant phosphorylation sites lack the consideration of kinase-specific phosphorylation data. Thus, we are motivated to propose a new method that investigates different substrate specificities in plant phosphorylation sites.
Experimentally verified phosphorylation data were extracted from TAIR9-a protein database containing 3006 phosphorylation data from the plant species Arabidopsis thaliana. In an attempt to investigate the various substrate motifs in plant phosphorylation, maximal dependence decomposition (MDD) is employed to cluster a large set of phosphorylation data into subgroups containing significantly conserved motifs. Profile hidden Markov model (HMM) is then applied to learn a predictive model for each subgroup. Cross-validation evaluation on the MDD-clustered HMMs yields an average accuracy of 82.4% for serine, 78.6% for threonine, and 89.0% for tyrosine models. Moreover, independent test results using Arabidopsis thaliana phosphorylation data from UniProtKB/Swiss-Prot show that the proposed models are able to correctly predict 81.4% phosphoserine, 77.1% phosphothreonine, and 83.7% phosphotyrosine sites. Interestingly, several MDD-clustered subgroups are observed to have similar amino acid conservation with the substrate motifs of well-known kinases from Phospho.ELM-a database containing kinase-specific phosphorylation data from multiple organisms.
This work presents a novel method for identifying plant phosphorylation sites with various substrate motifs. Based on cross-validation and independent testing, results show that the MDD-clustered models outperform models trained without using MDD. The proposed method has been implemented as a web-based plant phosphorylation prediction tool, PlantPhos http://csb.cse.yzu.edu.tw/PlantPhos/. Additionally, two case studies have been demonstrated to further evaluate the effectiveness of PlantPhos.
激酶催化的蛋白质磷酸化在细胞内信号转导中发挥着至关重要的调节作用。由于高通量质谱实验的难度,人们希望使用计算方法来预测磷酸化位点。然而,以前关于植物磷酸化位点的计算机预测研究缺乏对激酶特异性磷酸化数据的考虑。因此,我们有动力提出一种新的方法来研究植物磷酸化位点的不同底物特异性。
从包含拟南芥 3006 个磷酸化数据的 TAIR9 蛋白质数据库中提取了实验验证的磷酸化数据。为了研究植物磷酸化中的各种底物基序,我们采用最大依赖分解(MDD)将大量磷酸化数据聚类成包含显著保守基序的子组。然后应用轮廓隐马尔可夫模型(HMM)为每个子组学习预测模型。对 MDD 聚类的 HMM 进行交叉验证评估,得到丝氨酸模型的平均准确率为 82.4%,苏氨酸模型的准确率为 78.6%,酪氨酸模型的准确率为 89.0%。此外,使用 UniProtKB/Swiss-Prot 中的拟南芥磷酸化数据进行独立测试的结果表明,所提出的模型能够正确预测 81.4%的磷酸丝氨酸、77.1%的磷酸苏氨酸和 83.7%的磷酸酪氨酸位点。有趣的是,几个 MDD 聚类的子组被观察到与 Phospho.ELM-a 数据库中包含来自多个生物体的激酶特异性磷酸化数据的激酶特异性磷酸化数据的底物基序具有相似的氨基酸保守性。
本研究提出了一种识别具有不同底物基序的植物磷酸化位点的新方法。基于交叉验证和独立测试,结果表明,使用 MDD 聚类的模型优于不使用 MDD 聚类的模型。该方法已被实现为一个基于网络的植物磷酸化预测工具,PlantPhos http://csb.cse.yzu.edu.tw/PlantPhos/。此外,还进行了两个案例研究,以进一步评估 PlantPhos 的有效性。