College of Life Sciences and Systems Biology Research Center, Gwangju Institute of Science and Technology (GIST), Buk-gu, Gwangju, Republic of Korea.
PLoS One. 2012;7(8):e43282. doi: 10.1371/journal.pone.0043282. Epub 2012 Aug 28.
Histidine-rich calcium binding protein (HRC) is located in the lumen of sarcoplasmic reticulum (SR) that binds to both triadin (TRN) and SERCA affecting Ca(2+) cycling in the SR. Chronic overexpression of HRC that may disrupt intracellular Ca(2+) homeostasis is implicated in pathogenesis of cardiac hypertrophy. Ablation of HRC showed relatively normal phenotypes under basal condition, but exhibited a significantly increased susceptibility to isoproterenol-induced cardiac hypertrophy. In the present study, we characterized the functions of HRC related to Ca(2+) cycling and pathogenesis of cardiac hypertrophy using the in vitro siRNA- and the in vivo adeno-associated virus (AAV)-mediated HRC knock-down (KD) systems, respectively.
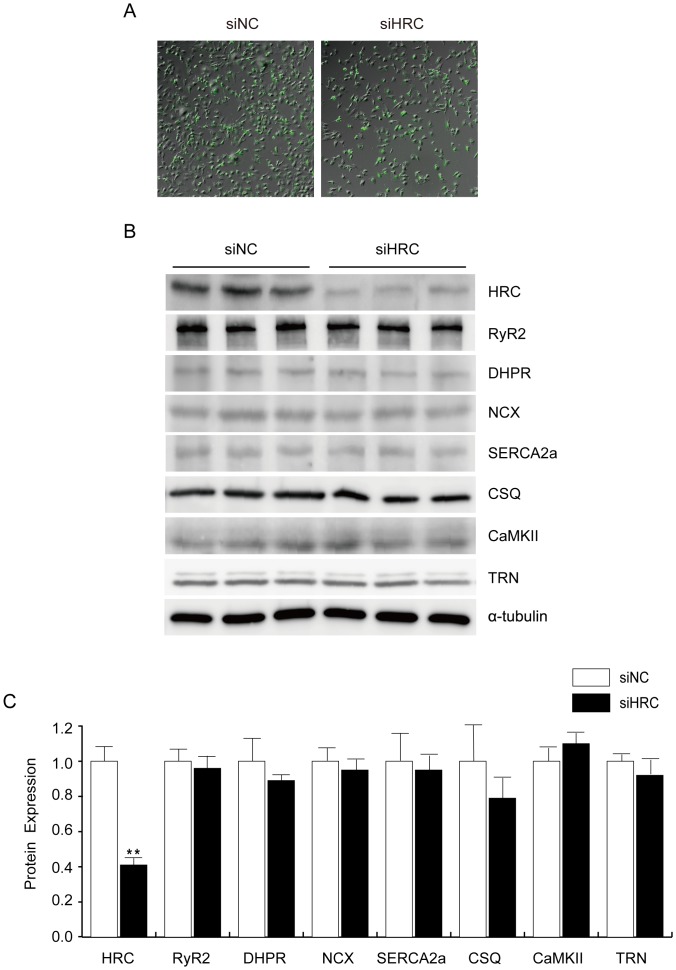
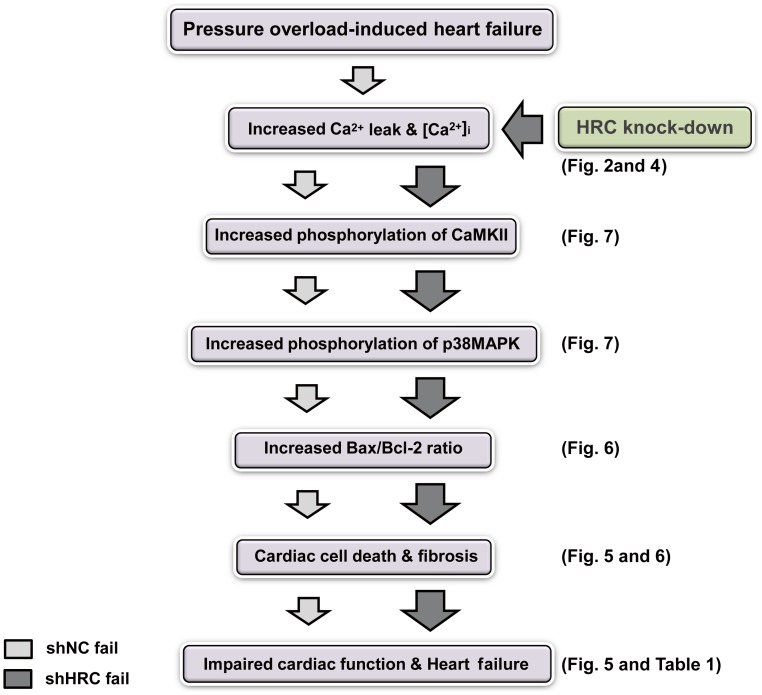
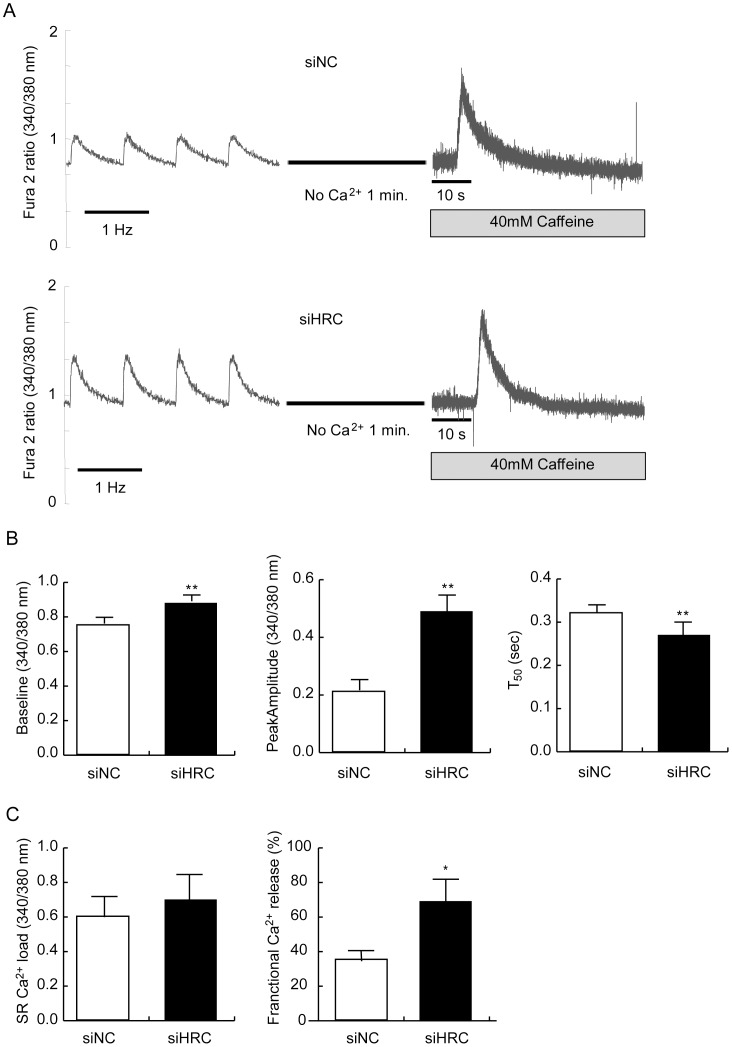
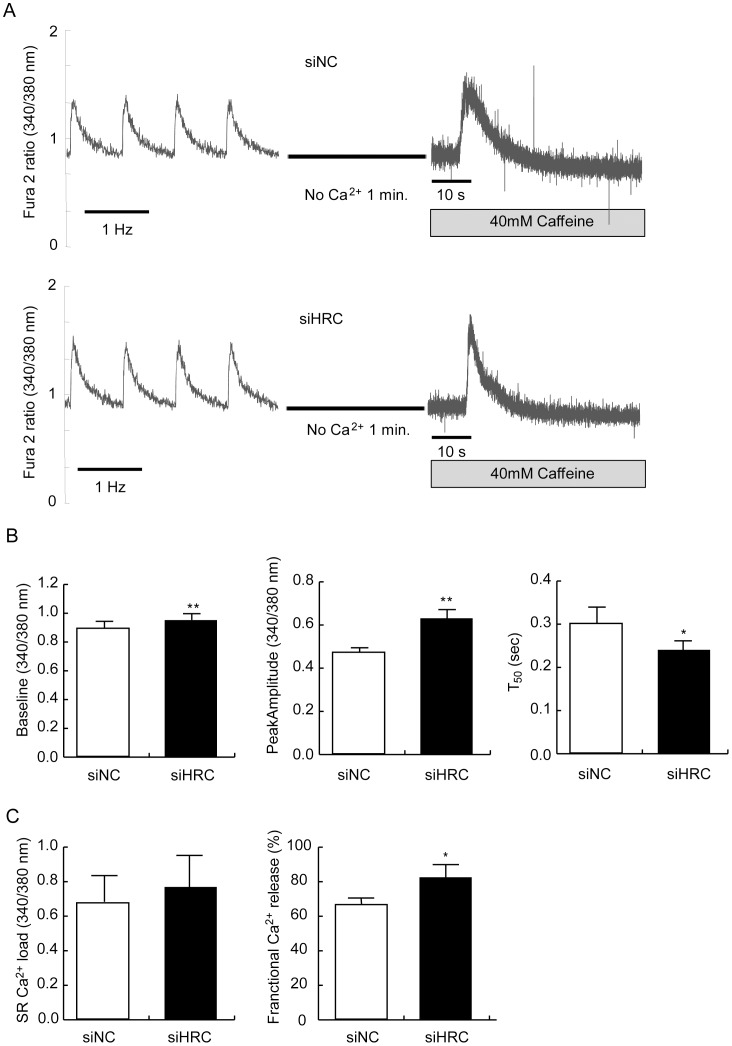
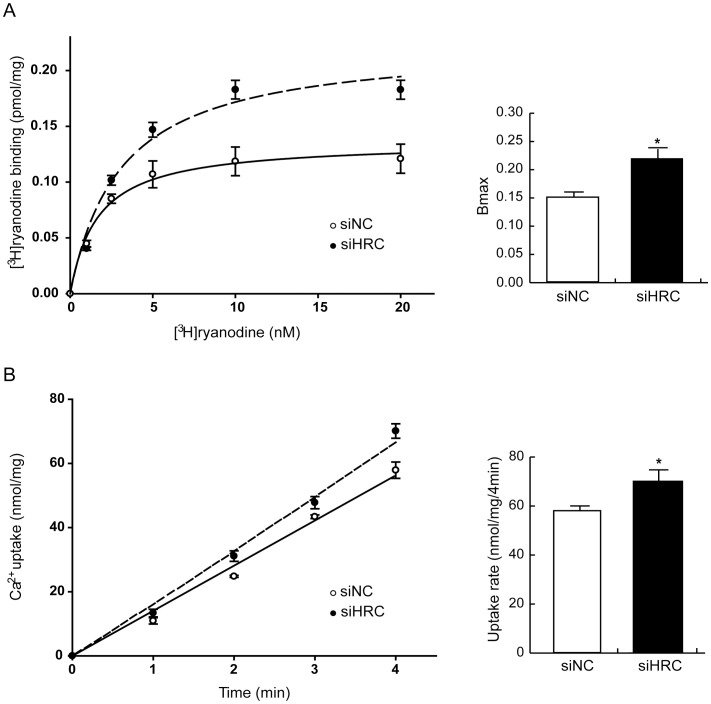
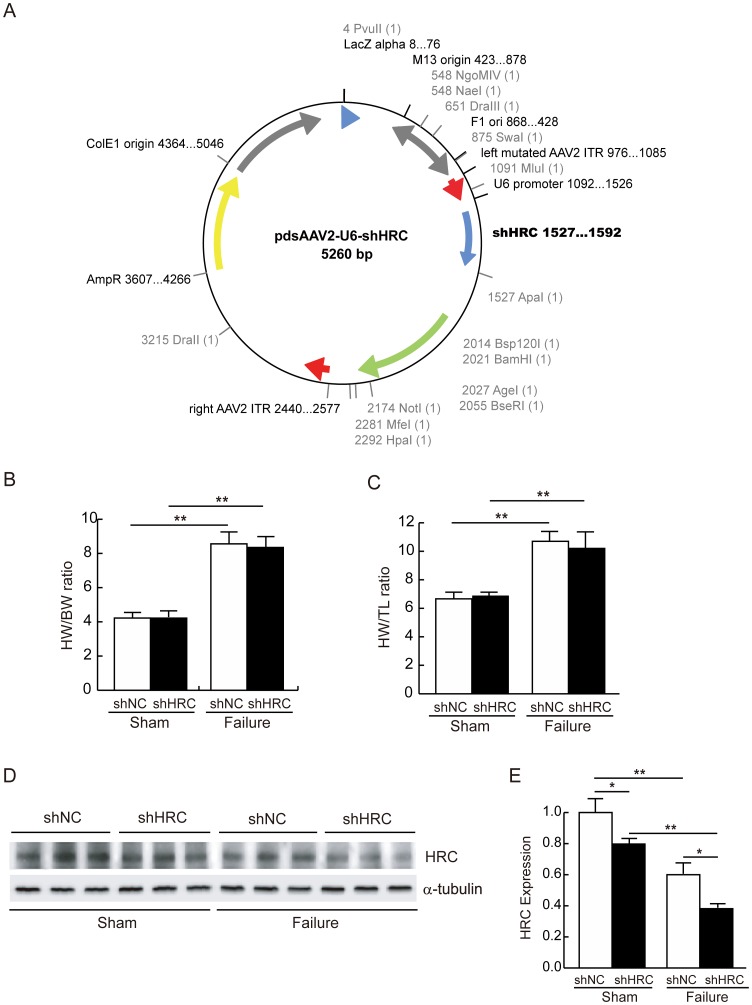
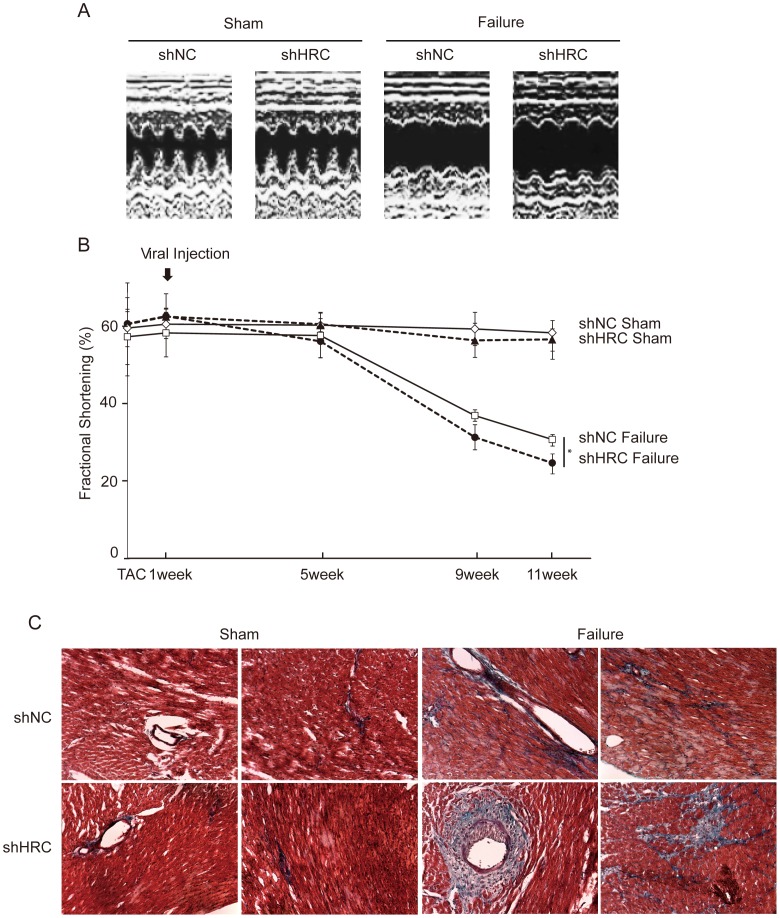
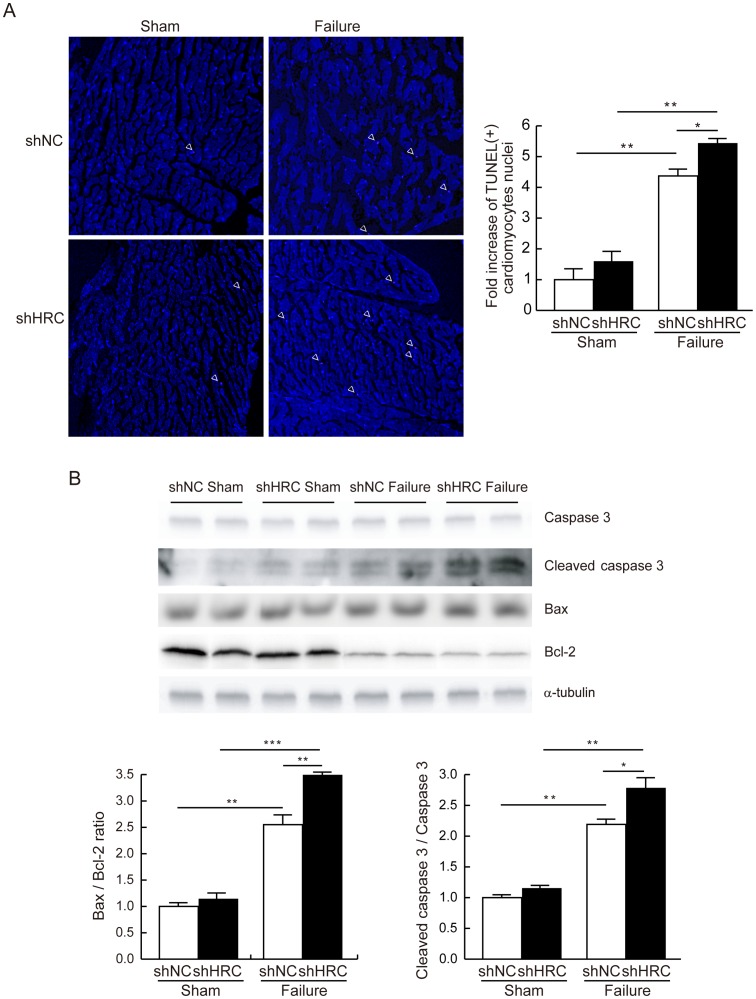
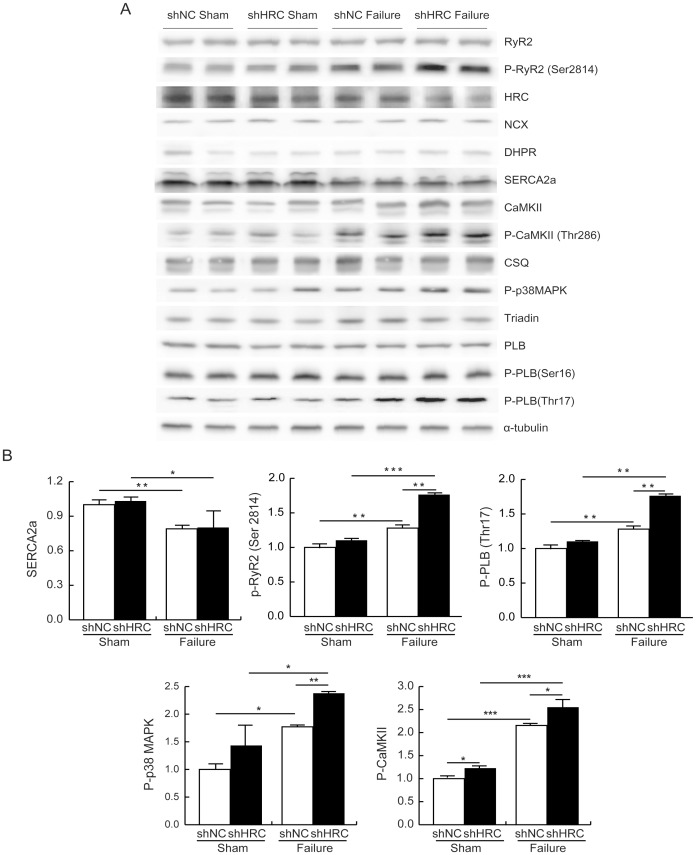
METHODOLOGY/PRINCIPAL FINDINGS: AAV-mediated HRC-KD system was used with or without C57BL/6 mouse model of transverse aortic constriction-induced failing heart (TAC-FH) to examine whether HRC-KD could enhance cardiac function in failing heart (FH). Initially we expected that HRC-KD could elicit cardiac functional recovery in failing heart (FH), since predesigned siRNA-mediated HRC-KD enhanced Ca(2+) cycling and increased activities of RyR2 and SERCA2 without change in SR Ca(2+) load in neonatal rat ventricular cells (NRVCs) and HL-1 cells. However, AAV9-mediated HRC-KD in TAC-FH was associated with decreased fractional shortening and increased cardiac fibrosis compared with control. We found that phospho-RyR2, phospho-CaMKII, phospho-p38 MAPK, and phospho-PLB were significantly upregulated by HRC-KD in TAC-FH. A significantly increased level of cleaved caspase-3, a cardiac cell death marker was also found, consistent with the result of TUNEL assay.
CONCLUSIONS/SIGNIFICANCE: Increased Ca(2+) leak and cytosolic Ca(2+) concentration due to a partial KD of HRC could enhance activity of CaMKII and phosphorylation of p38 MAPK, causing the mitochondrial death pathway observed in TAC-FH. Our results present evidence that down-regulation of HRC could deteriorate cardiac function in TAC-FH through perturbed SR-mediated Ca(2+) cycling.
组氨酸丰富的钙结合蛋白(HRC)位于肌浆网(SR)的腔中,与连接蛋白(TRN)和 SERCA 结合,影响 SR 中的 Ca(2+)循环。慢性 HRC 过度表达可能破坏细胞内 Ca(2+)稳态,与心肌肥厚的发病机制有关。HRC 缺失在基础条件下表现出相对正常的表型,但对异丙肾上腺素诱导的心肌肥厚表现出明显增加的易感性。在本研究中,我们使用体外 siRNA 和体内腺相关病毒(AAV)介导的 HRC 敲低(KD)系统,分别表征与 Ca(2+)循环和心肌肥厚发病机制相关的 HRC 功能。
方法/主要发现:使用 AAV 介导的 HRC-KD 系统,结合或不结合 C57BL/6 小鼠的主动脉缩窄诱导心力衰竭(TAC-FH)模型,以检查 HRC-KD 是否可以增强心力衰竭(FH)中的心脏功能。最初,我们期望 HRC-KD 可以引起 FH 中的心脏功能恢复,因为预先设计的 siRNA 介导的 HRC-KD 增强了 Ca(2+)循环,增加了 RyR2 和 SERCA2 的活性,而在新生大鼠心室细胞(NRVCs)和 HL-1 细胞中 SR Ca(2+)负荷没有变化。然而,与对照相比,AAV9 介导的 TAC-FH 中的 HRC-KD 与缩短分数降低和心脏纤维化增加有关。我们发现,在 TAC-FH 中,HRC-KD 显著上调了磷酸化 RyR2、磷酸化 CaMKII、磷酸化 p38 MAPK 和磷酸化 PLB。还发现一种心脏细胞死亡标志物 cleaved caspase-3 的水平显著增加,与 TUNEL 测定的结果一致。
结论/意义:由于 HRC 的部分 KD,Ca(2+)泄漏和细胞溶质 Ca(2+)浓度增加,可能会增强 CaMKII 的活性和 p38 MAPK 的磷酸化,导致在 TAC-FH 中观察到的线粒体死亡途径。我们的结果提供了证据,表明在 TAC-FH 中,HRC 的下调可能通过扰乱 SR 介导的 Ca(2+)循环而使心脏功能恶化。