Department of Genome Sciences, University of Washington, Seattle, Washington 98195, USA.
Genome Res. 2012 Sep;22(9):1680-8. doi: 10.1101/gr.136101.111.
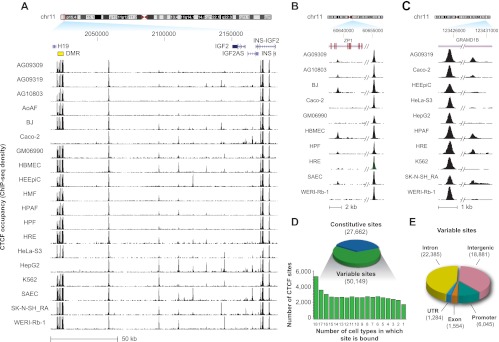
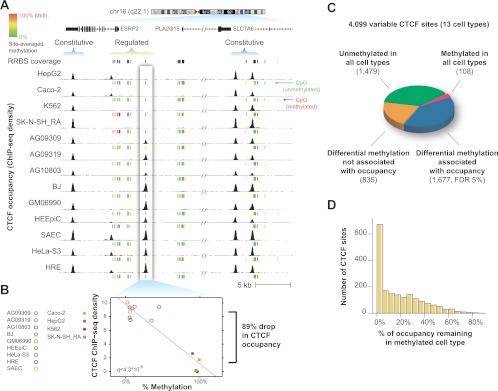
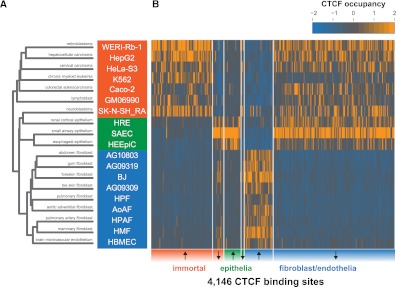
CTCF is a ubiquitously expressed regulator of fundamental genomic processes including transcription, intra- and interchromosomal interactions, and chromatin structure. Because of its critical role in genome function, CTCF binding patterns have long been assumed to be largely invariant across different cellular environments. Here we analyze genome-wide occupancy patterns of CTCF by ChIP-seq in 19 diverse human cell types, including normal primary cells and immortal lines. We observed highly reproducible yet surprisingly plastic genomic binding landscapes, indicative of strong cell-selective regulation of CTCF occupancy. Comparison with massively parallel bisulfite sequencing data indicates that 41% of variable CTCF binding is linked to differential DNA methylation, concentrated at two critical positions within the CTCF recognition sequence. Unexpectedly, CTCF binding patterns were markedly different in normal versus immortal cells, with the latter showing widespread disruption of CTCF binding associated with increased methylation. Strikingly, this disruption is accompanied by up-regulation of CTCF expression, with the result that both normal and immortal cells maintain the same average number of CTCF occupancy sites genome-wide. These results reveal a tight linkage between DNA methylation and the global occupancy patterns of a major sequence-specific regulatory factor.
CTCF 是一种普遍表达的调节因子,参与多种基本的基因组过程,包括转录、染色体内和染色体间相互作用以及染色质结构。由于其在基因组功能中的关键作用,CTCF 的结合模式长期以来被认为在不同的细胞环境中基本保持不变。在这里,我们通过 ChIP-seq 分析了 19 种不同的人类细胞类型(包括正常原代细胞和永生化细胞系)中 CTCF 的全基因组占据模式。我们观察到高度可重复但令人惊讶的灵活基因组结合景观,表明 CTCF 占据的强烈细胞选择性调节。与大规模平行亚硫酸氢盐测序数据的比较表明,41%的可变 CTCF 结合与差异 DNA 甲基化有关,集中在 CTCF 识别序列内的两个关键位置。出乎意料的是,正常细胞与永生化细胞之间的 CTCF 结合模式明显不同,后者显示 CTCF 结合广泛中断,伴随着甲基化增加。引人注目的是,这种中断伴随着 CTCF 表达的上调,结果是正常细胞和永生化细胞在全基因组范围内保持相同数量的 CTCF 占据位点。这些结果揭示了 DNA 甲基化和主要序列特异性调节因子的全局占据模式之间的紧密联系。