Department of Genetics, Stanford University School of Medicine, Stanford, California 94305, USA.
Genome Res. 2012 Sep;22(9):1790-7. doi: 10.1101/gr.137323.112.
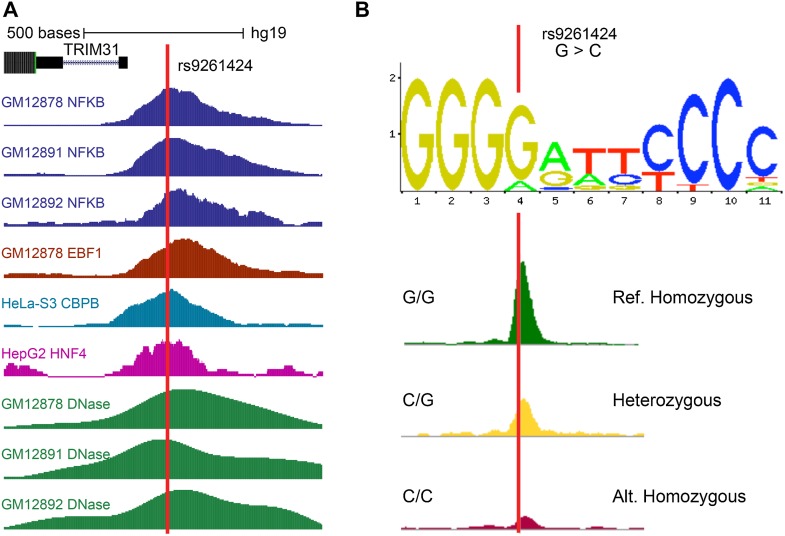
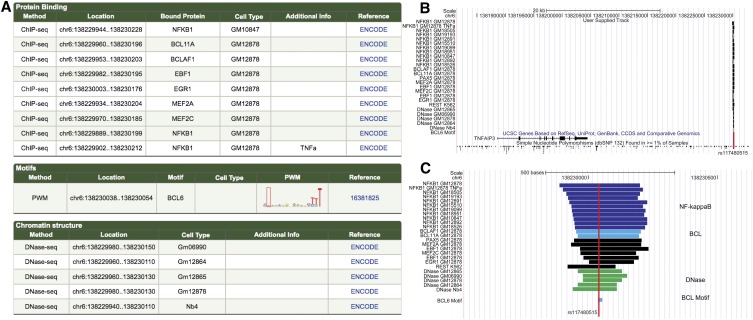
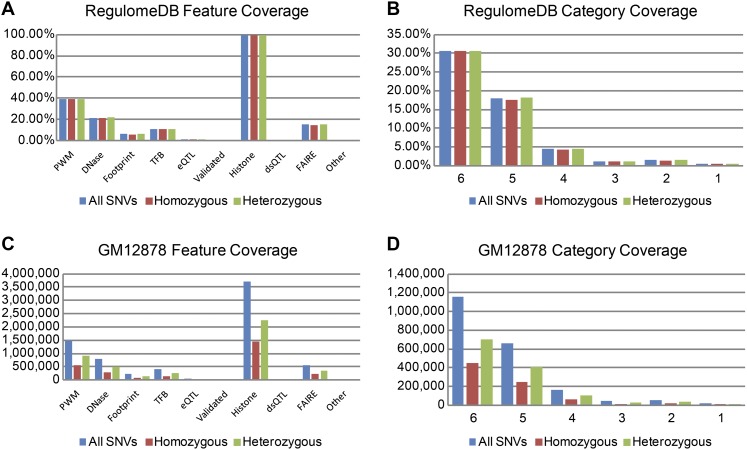
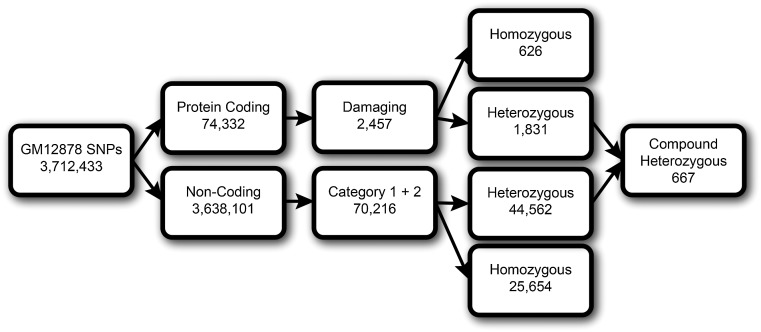
As the sequencing of healthy and disease genomes becomes more commonplace, detailed annotation provides interpretation for individual variation responsible for normal and disease phenotypes. Current approaches focus on direct changes in protein coding genes, particularly nonsynonymous mutations that directly affect the gene product. However, most individual variation occurs outside of genes and, indeed, most markers generated from genome-wide association studies (GWAS) identify variants outside of coding segments. Identification of potential regulatory changes that perturb these sites will lead to a better localization of truly functional variants and interpretation of their effects. We have developed a novel approach and database, RegulomeDB, which guides interpretation of regulatory variants in the human genome. RegulomeDB includes high-throughput, experimental data sets from ENCODE and other sources, as well as computational predictions and manual annotations to identify putative regulatory potential and identify functional variants. These data sources are combined into a powerful tool that scores variants to help separate functional variants from a large pool and provides a small set of putative sites with testable hypotheses as to their function. We demonstrate the applicability of this tool to the annotation of noncoding variants from 69 full sequenced genomes as well as that of a personal genome, where thousands of functionally associated variants were identified. Moreover, we demonstrate a GWAS where the database is able to quickly identify the known associated functional variant and provide a hypothesis as to its function. Overall, we expect this approach and resource to be valuable for the annotation of human genome sequences.
随着健康和疾病基因组测序变得越来越普遍,详细的注释为负责正常和疾病表型的个体变异提供了解释。目前的方法主要集中在蛋白质编码基因的直接变化上,特别是直接影响基因产物的非同义突变。然而,大多数个体变异发生在基因之外,事实上,大多数来自全基因组关联研究(GWAS)的标记都识别出编码片段之外的变体。鉴定可能扰乱这些位点的潜在调控变化,将导致更准确地定位真正的功能变体,并解释其影响。我们开发了一种新的方法和数据库,RegulomeDB,用于指导人类基因组中调控变体的解释。RegulomeDB 包括来自 ENCODE 和其他来源的高通量、实验数据集,以及计算预测和手动注释,以识别潜在的调控潜力和鉴定功能变体。这些数据源被组合成一个强大的工具,对变体进行评分,帮助将功能变体与大量变体区分开来,并提供一小部分具有可测试假设的假定位点,以了解其功能。我们展示了该工具在注释 69 个全测序基因组和个人基因组中非编码变体的适用性,在这两个基因组中鉴定出了数千个功能相关的变体。此外,我们还展示了一个 GWAS,该数据库能够快速识别已知相关的功能变体,并提供其功能的假设。总的来说,我们期望这种方法和资源对人类基因组序列的注释具有重要价值。