Department of Human Genetics, University of Chicago, Chicago, Illinois 60637, USA.
Nature. 2012 Feb 5;482(7385):390-4. doi: 10.1038/nature10808.
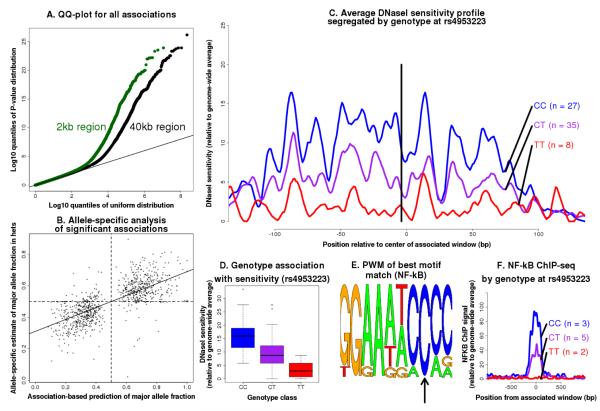
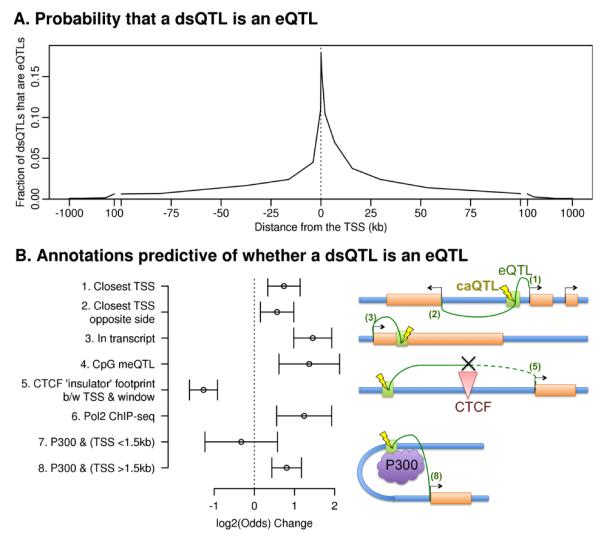
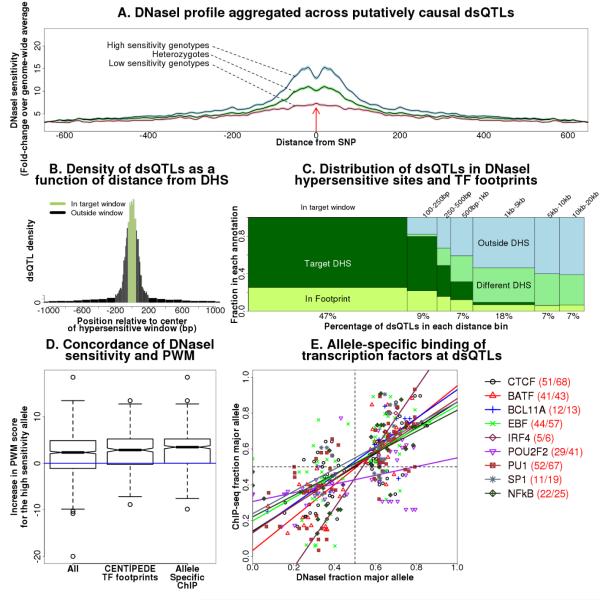
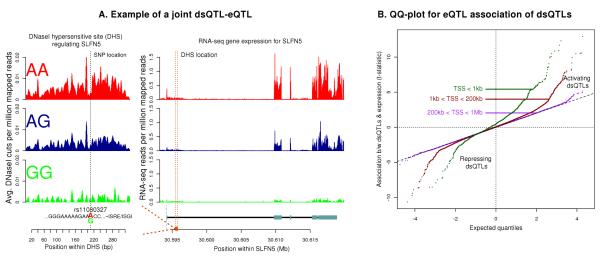
The mapping of expression quantitative trait loci (eQTLs) has emerged as an important tool for linking genetic variation to changes in gene regulation. However, it remains difficult to identify the causal variants underlying eQTLs, and little is known about the regulatory mechanisms by which they act. Here we show that genetic variants that modify chromatin accessibility and transcription factor binding are a major mechanism through which genetic variation leads to gene expression differences among humans. We used DNase I sequencing to measure chromatin accessibility in 70 Yoruba lymphoblastoid cell lines, for which genome-wide genotypes and estimates of gene expression levels are also available. We obtained a total of 2.7 billion uniquely mapped DNase I-sequencing (DNase-seq) reads, which allowed us to produce genome-wide maps of chromatin accessibility for each individual. We identified 8,902 locations at which the DNase-seq read depth correlated significantly with genotype at a nearby single nucleotide polymorphism or insertion/deletion (false discovery rate = 10%). We call such variants 'DNase I sensitivity quantitative trait loci' (dsQTLs). We found that dsQTLs are strongly enriched within inferred transcription factor binding sites and are frequently associated with allele-specific changes in transcription factor binding. A substantial fraction (16%) of dsQTLs are also associated with variation in the expression levels of nearby genes (that is, these loci are also classified as eQTLs). Conversely, we estimate that as many as 55% of eQTL single nucleotide polymorphisms are also dsQTLs. Our observations indicate that dsQTLs are highly abundant in the human genome and are likely to be important contributors to phenotypic variation.
表达数量性状基因座(eQTL)的图谱绘制已成为将遗传变异与基因调控变化联系起来的重要工具。然而,确定 eQTL 背后的因果变异仍然很困难,并且人们对它们起作用的调节机制知之甚少。在这里,我们表明,改变染色质可及性和转录因子结合的遗传变异是遗传变异导致人类基因表达差异的主要机制。我们使用 DNaseI 测序来测量 70 个约鲁巴淋巴母细胞系中的染色质可及性,这些细胞系还具有全基因组基因型和基因表达水平的估计值。我们总共获得了 27 亿个唯一映射的 DNaseI 测序(DNase-seq)读取,这使我们能够为每个个体生成全基因组染色质可及性图谱。我们确定了 8902 个位置,在这些位置上,DNase-seq 读取深度与附近单核苷酸多态性或插入/缺失的基因型显著相关(错误发现率 = 10%)。我们将此类变体称为“DNaseI 敏感性数量性状基因座”(dsQTL)。我们发现,dsQTL 在内推断转录因子结合位点中强烈富集,并且经常与转录因子结合的等位基因特异性变化相关联。相当一部分(16%)dsQTL 也与附近基因表达水平的变异相关(即,这些位点也被归类为 eQTL)。相反,我们估计多达 55%的 eQTL 单核苷酸多态性也是 dsQTL。我们的观察表明,dsQTL 在人类基因组中非常丰富,并且可能是表型变异的重要贡献者。