Aviram Iddo, Veltman Ilia, Churkin Alexander, Barash Danny
Department of Computer Science, Ben-Gurion University, 84105, Beer Sheva, Israel.
Algorithms Mol Biol. 2012 Sep 7;7(1):24. doi: 10.1186/1748-7188-7-24.
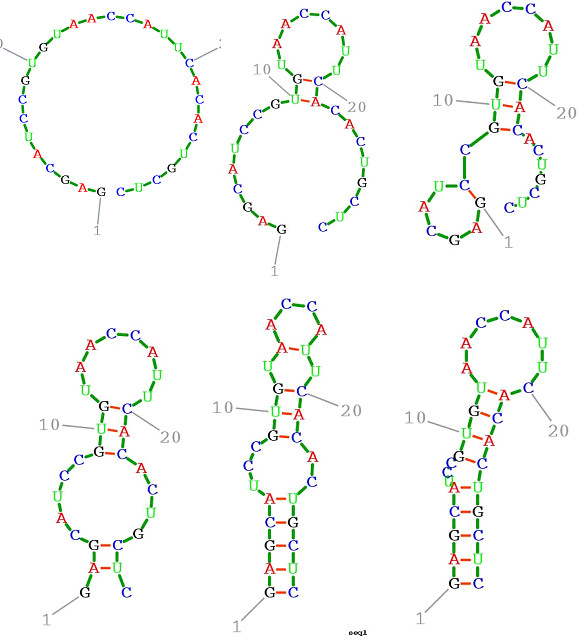
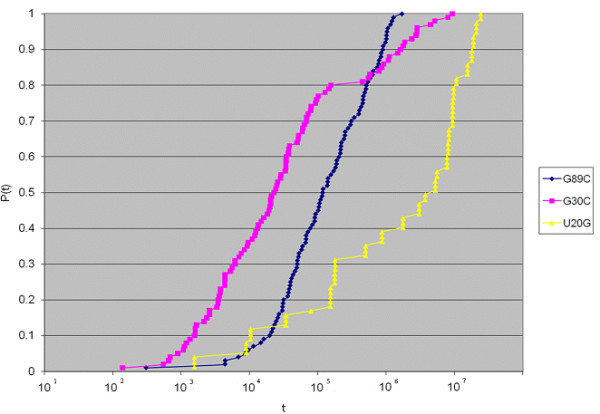
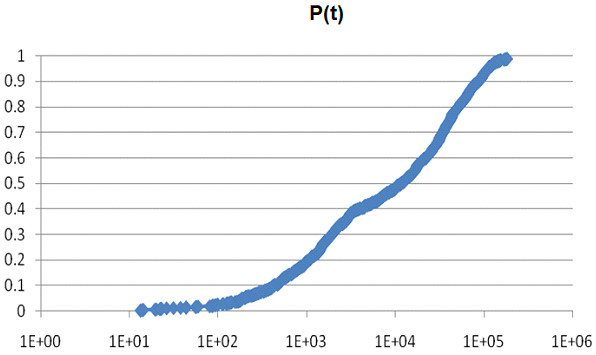
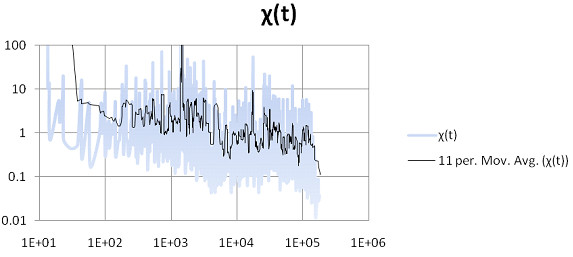
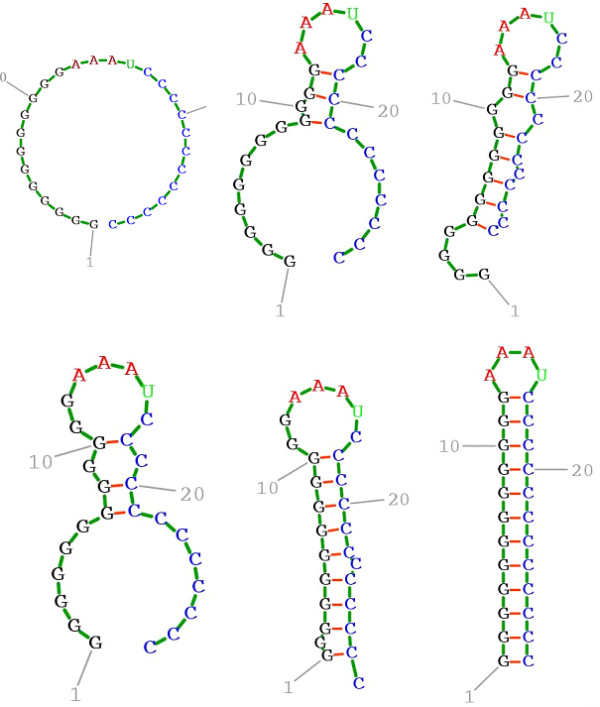
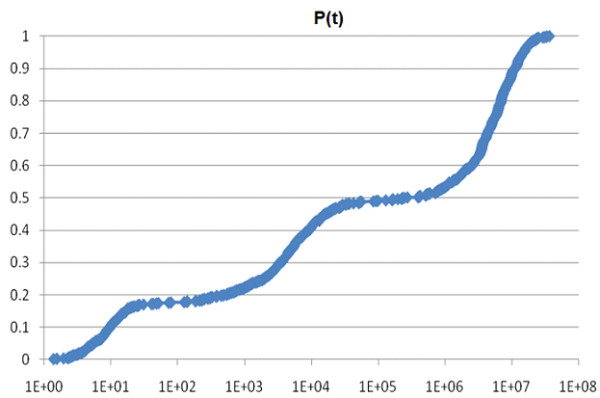
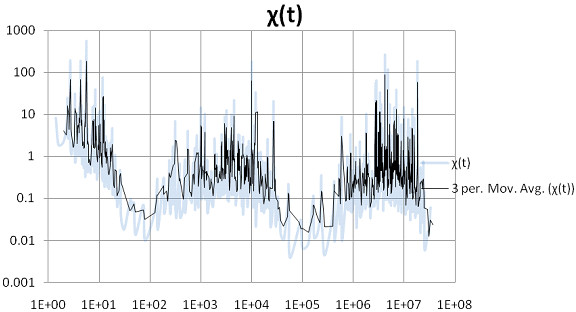
Methods for simulating the kinetic folding of RNAs by numerically solving the chemical master equation have been developed since the late 90's, notably the programs Kinfold and Treekin with Barriers that are available in the Vienna RNA package. Our goal is to formulate extensions to the algorithms used, starting from the Gillespie algorithm, that will allow numerical simulations of mid-size (~ 60-150 nt) RNA kinetics in some practical cases where numerous distributions of folding times are desired. These extensions can contribute to analyses and predictions of RNA folding in biologically significant problems.
By describing in a particular way the reduction of numerical simulations of RNA folding kinetics into the Gillespie stochastic simulation algorithm for chemical reactions, it is possible to formulate extensions to the basic algorithm that will exploit memoization and parallelism for efficient computations. These can be used to advance forward from the small examples demonstrated to larger examples of biological interest.
The implementation that is described and used for the Gillespie algorithm is freely available by contacting the authors, noting that the efficient procedures suggested may also be applicable along with Vienna's Kinfold.
自90年代末以来,已经开发了通过数值求解化学主方程来模拟RNA动力学折叠的方法,特别是维也纳RNA软件包中的Kinfold程序和带有障碍的Treekin程序。我们的目标是从 Gillespie算法开始,对所用算法进行扩展,以便在一些需要多种折叠时间分布的实际情况下,能够对中等大小(约60 - 150个核苷酸)的RNA动力学进行数值模拟。这些扩展有助于在具有生物学意义的问题中对RNA折叠进行分析和预测。
通过以一种特定方式将RNA折叠动力学的数值模拟简化为化学反应的Gillespie随机模拟算法,可以对基本算法进行扩展,利用记忆化和并行性进行高效计算。这些扩展可用于从已展示的小示例推进到更具生物学意义的大示例。
通过联系作者可免费获得所描述并用于Gillespie算法的实现,请注意所建议的高效程序也可与维也纳的Kinfold一起应用。