Department of Otorhinolaryngology, 1P1, Ghent University / Ghent University Hospital, De Pintelaan 185, Ghent 9000, Belgium.
Orphanet J Rare Dis. 2012 Oct 30;7:84. doi: 10.1186/1750-1172-7-84.
Stickler syndrome is a connective tissue disorder characterized by ocular, skeletal, orofacial and auditory defects. It is caused by mutations in different collagen genes, namely COL2A1, COL11A1 and COL11A2 (autosomal dominant inheritance), and COL9A1 and COL9A2 (autosomal recessive inheritance). The auditory phenotype in Stickler syndrome is inconsistently reported. Therefore we performed a systematic review of the literature to give an up-to-date overview of hearing loss in Stickler syndrome, and correlated it with the genotype.
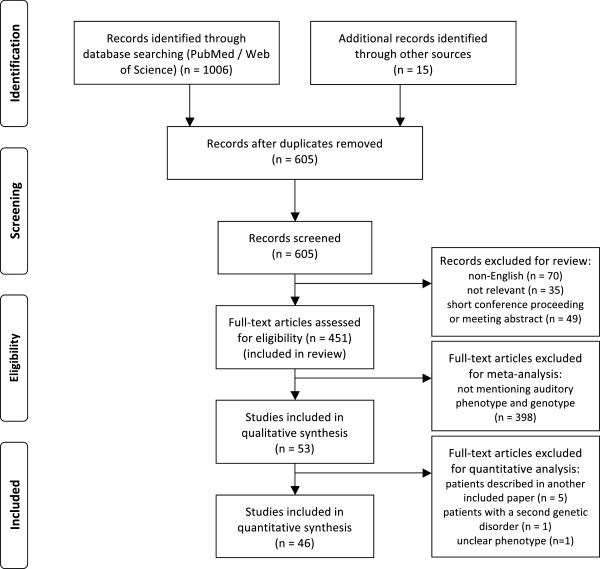
English-language literature was reviewed through searches of PubMed and Web of Science, in order to find relevant articles describing auditory features in Stickler patients, along with genotype. Prevalences of hearing loss are calculated and correlated with the different affected genes and type of mutation.
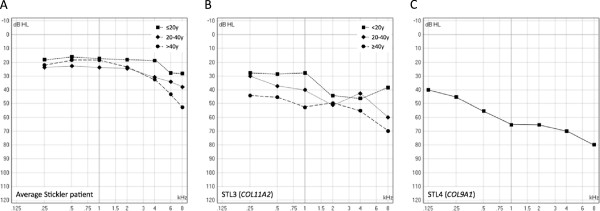
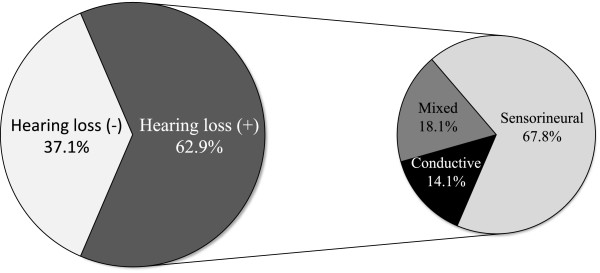
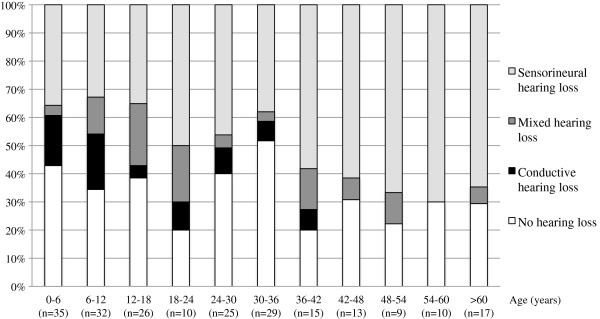
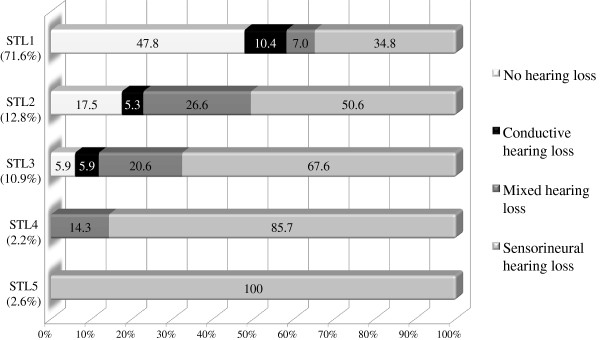
313 patients (102 families) individually described in 46 articles were included. Hearing loss was found in 62.9%, mostly mild to moderate when reported. Hearing impairment was predominantly sensorineural (67.8%). Conductive (14.1%) and mixed (18.1%) hearing loss was primarily found in young patients or patients with a palatal defect. Overall, mutations in COL11A1 (82.5%) and COL11A2 (94.1%) seem to be more frequently associated with hearing impairment than mutations in COL2A1 (52.2%).
Hearing impairment in patients with Stickler syndrome is common. Sensorineural hearing loss predominates, but also conductive hearing loss, especially in children and patients with a palatal defect, may occur. The distinct disease-causing collagen genes are associated with a different prevalence of hearing impairment, but still large phenotypic variation exists. Regular auditory follow-up is strongly advised, particularly because many Stickler patients are visually impaired.
斯特格勒综合征是一种结缔组织疾病,其特征为眼部、骨骼、口腔面和听觉缺陷。它是由不同的胶原基因,即 COL2A1、COL11A1 和 COL11A2(常染色体显性遗传)以及 COL9A1 和 COL9A2(常染色体隐性遗传)突变引起的。斯特格勒综合征的听觉表型报道不一致。因此,我们进行了系统的文献回顾,以提供有关斯特格勒综合征听力损失的最新概述,并将其与基因型相关联。
通过在 PubMed 和 Web of Science 上进行英语文献检索,回顾了相关文献,以找到描述斯特格勒患者听觉特征以及基因型的文章。计算听力损失的患病率,并与不同受影响的基因和突变类型相关联。
纳入了 46 篇文章中单独描述的 313 名患者(102 个家庭)。听力损失发生率为 62.9%,报告中多为轻度至中度。听力障碍主要为感音神经性(67.8%)。传导性(14.1%)和混合性(18.1%)听力障碍主要发生在年轻患者或有腭裂缺陷的患者中。总体而言,COL11A1(82.5%)和 COL11A2(94.1%)突变似乎比 COL2A1(52.2%)突变更常与听力障碍相关。
斯特格勒综合征患者的听力障碍很常见。感音神经性听力损失为主,但也可能发生传导性听力损失,尤其是在儿童和有腭裂缺陷的患者中。不同的致病胶原基因与听力障碍的不同患病率相关,但仍存在较大的表型变异。强烈建议进行定期的听觉随访,特别是因为许多斯特格勒综合征患者存在视力障碍。