Laboratorio de Virología Molecular, Centro de Investigaciones Nucleares, Facultad de Ciencias, Universidad de la República, Iguá 4225, Montevideo, 11400, Uruguay.
Virol J. 2012 Nov 8;9:263. doi: 10.1186/1743-422X-9-263.
Influenza A virus (IAV) is a member of the family Orthomyxoviridae and contains eight segments of a single-stranded RNA genome with negative polarity. The first influenza pandemic of this century was declared in April of 2009, with the emergence of a novel H1N1 IAV strain (H1N1pdm) in Mexico and USA. Understanding the extent and causes of biases in codon usage is essential to the understanding of viral evolution. A comprehensive study to investigate the effect of selection pressure imposed by the human host on the codon usage of an emerging, pandemic IAV strain and the trends in viral codon usage involved over the pandemic time period is much needed.
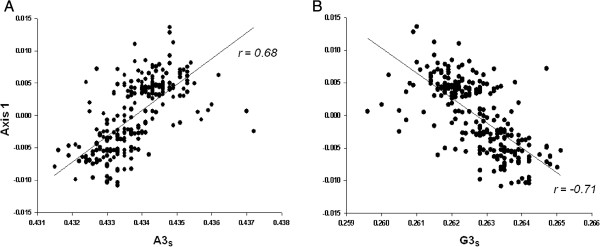
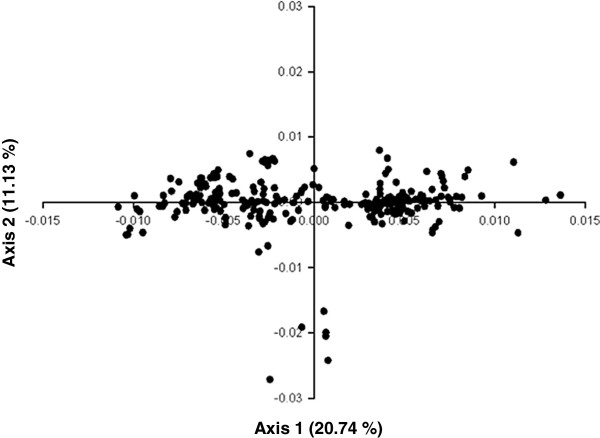
We performed a comprehensive codon usage analysis of 310 IAV strains from the pandemic of 2009. Highly biased codon usage for Ala, Arg, Pro, Thr and Ser were found. Codon usage is strongly influenced by underlying biases in base composition. When correspondence analysis (COA) on relative synonymous codon usage (RSCU) is applied, the distribution of IAV ORFs in the plane defined by the first two major dimensional factors showed that different strains are located at different places, suggesting that IAV codon usage also reflects an evolutionary process.
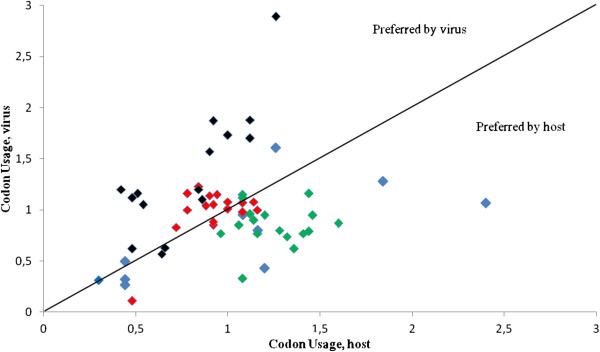
A general association between codon usage bias, base composition and poor adaptation of the virus to the respective host tRNA pool, suggests that mutational pressure is the main force shaping H1N1 pdm IAV codon usage. A dynamic process is observed in the variation of codon usage of the strains enrolled in these studies. These results suggest a balance of mutational bias and natural selection, which allow the virus to explore and re-adapt its codon usage to different environments. Recoding of IAV taking into account codon bias, base composition and adaptation to host tRNA may provide important clues to develop new and appropriate vaccines.
甲型流感病毒(IAV)是正黏病毒科的一个成员,包含 8 个单链 RNA 基因组片段,具有负极性。本世纪的第一次流感大流行于 2009 年 4 月宣布,在墨西哥和美国出现了一种新型 H1N1 IAV 株(H1N1pdm)。了解密码子使用偏倚的程度和原因对于理解病毒进化至关重要。非常需要对选择压力对新兴大流行 IAV 株的密码子使用的影响以及大流行期间病毒密码子使用趋势进行全面研究。
我们对 2009 年大流行期间的 310 株 IAV 株进行了全面的密码子使用分析。发现 Ala、Arg、Pro、Thr 和 Ser 的密码子使用高度偏倚。密码子使用受到碱基组成的潜在偏倚的强烈影响。当在相对同义密码子使用(RSCU)上应用对应分析(COA)时,IAV ORF 在由前两个主要维度因素定义的平面上的分布表明,不同的菌株位于不同的位置,这表明 IAV 密码子使用也反映了一个进化过程。
密码子使用偏倚、碱基组成和病毒对各自宿主 tRNA 池的适应不良之间的一般关联表明,突变压力是塑造 H1N1pdm IAV 密码子使用的主要力量。在这些研究中登记的菌株的密码子使用变化中观察到一个动态过程。这些结果表明突变偏差和自然选择之间的平衡,使病毒能够探索并重新适应其密码子使用以适应不同的环境。考虑到密码子偏倚、碱基组成和对宿主 tRNA 的适应,对 IAV 的重编码可能为开发新的和适当的疫苗提供重要线索。