Department of Entomology, State Museum of Natural History, Stuttgart, Germany.
PLoS One. 2012;7(11):e47450. doi: 10.1371/journal.pone.0047450. Epub 2012 Nov 20.
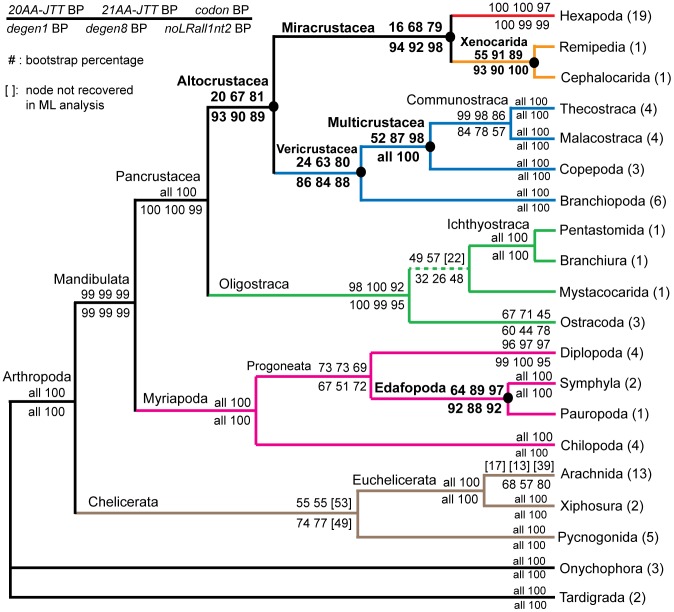
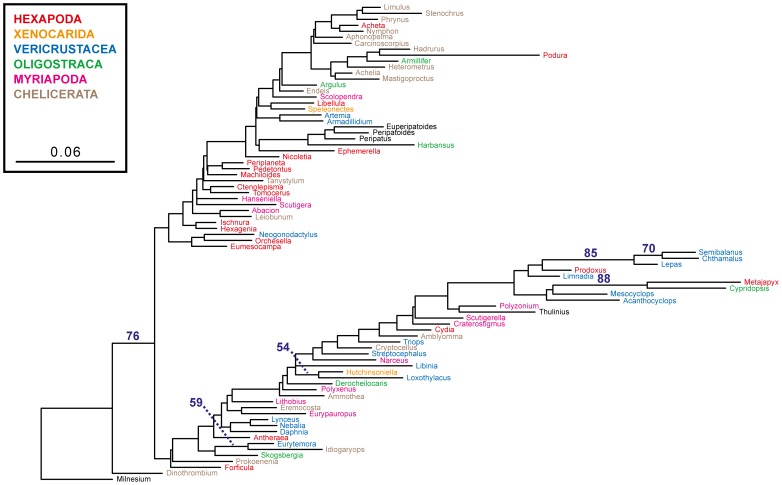
In a previous study of higher-level arthropod phylogeny, analyses of nucleotide sequences from 62 protein-coding nuclear genes for 80 panarthopod species yielded significantly higher bootstrap support for selected nodes than did amino acids. This study investigates the cause of that discrepancy.
METHODOLOGY/PRINCIPAL FINDINGS: The hypothesis is tested that failure to distinguish the serine residues encoded by two disjunct clusters of codons (TCN, AGY) in amino acid analyses leads to this discrepancy. In one test, the two clusters of serine codons (Ser1, Ser2) are conceptually translated as separate amino acids. Analysis of the resulting 21-amino-acid data matrix shows striking increases in bootstrap support, in some cases matching that in nucleotide analyses. In a second approach, nucleotide and 20-amino-acid data sets are artificially altered through targeted deletions, modifications, and replacements, revealing the pivotal contributions of distinct Ser1 and Ser2 codons. We confirm that previous methods of coding nonsynonymous nucleotide change are robust and computationally efficient by introducing two new degeneracy coding methods. We demonstrate for degeneracy coding that neither compositional heterogeneity at the level of nucleotides nor codon usage bias between Ser1 and Ser2 clusters of codons (or their separately coded amino acids) is a major source of non-phylogenetic signal.
The incongruity in support between amino-acid and nucleotide analyses of the forementioned arthropod data set is resolved by showing that "standard" 20-amino-acid analyses yield lower node support specifically when serine provides crucial signal. Separate coding of Ser1 and Ser2 residues yields support commensurate with that found by degenerated nucleotides, without introducing phylogenetic artifacts. While exclusion of all serine data leads to reduced support for serine-sensitive nodes, these nodes are still recovered in the ML topology, indicating that the enhanced signal from Ser1 and Ser2 is not qualitatively different from that of the other amino acids.
在之前的高级节肢动物系统发育研究中,对 80 种泛节肢动物物种的 62 个核蛋白编码基因的核苷酸序列进行分析,结果显示,与氨基酸分析相比,所选节点的自举支持率显著提高。本研究探讨了这种差异的原因。
方法/主要发现:假设在氨基酸分析中未能区分两个不连续的丝氨酸密码子簇(TCN、AGY)编码的丝氨酸残基导致了这种差异。在一项测试中,两个丝氨酸密码子簇(Ser1、Ser2)被概念上翻译为单独的氨基酸。分析得到的 21 个氨基酸数据矩阵显示,自举支持率显著增加,在某些情况下与核苷酸分析相匹配。在第二种方法中,通过有针对性的缺失、修改和替换,人为地改变核苷酸和 20 个氨基酸数据集,揭示了不同的 Ser1 和 Ser2 密码子的关键贡献。我们通过引入两种新的简并编码方法,证实了以前的编码非同义核苷酸变化的方法是稳健和计算有效的。我们证明,对于简并编码,核苷酸水平的组成异质性或 Ser1 和 Ser2 密码子簇(或它们分别编码的氨基酸)之间的密码子使用偏好都不是非系统发育信号的主要来源。
通过显示“标准”20 个氨基酸分析在丝氨酸提供关键信号时特异性地产生较低的节点支持,解决了前面提到的节肢动物数据集的氨基酸和核苷酸分析之间的不一致性。Ser1 和 Ser2 残基的单独编码产生与退化核苷酸相同的支持,而不会引入系统发育伪影。虽然排除所有丝氨酸数据会降低对丝氨酸敏感节点的支持,但这些节点仍然在 ML 拓扑结构中恢复,这表明来自 Ser1 和 Ser2 的增强信号与其他氨基酸的信号在质量上没有区别。