Department of Pharmaceutical Chemistry, University of California San Francisco, San Francisco, California, United States of America.
PLoS One. 2012;7(11):e49910. doi: 10.1371/journal.pone.0049910. Epub 2012 Nov 21.
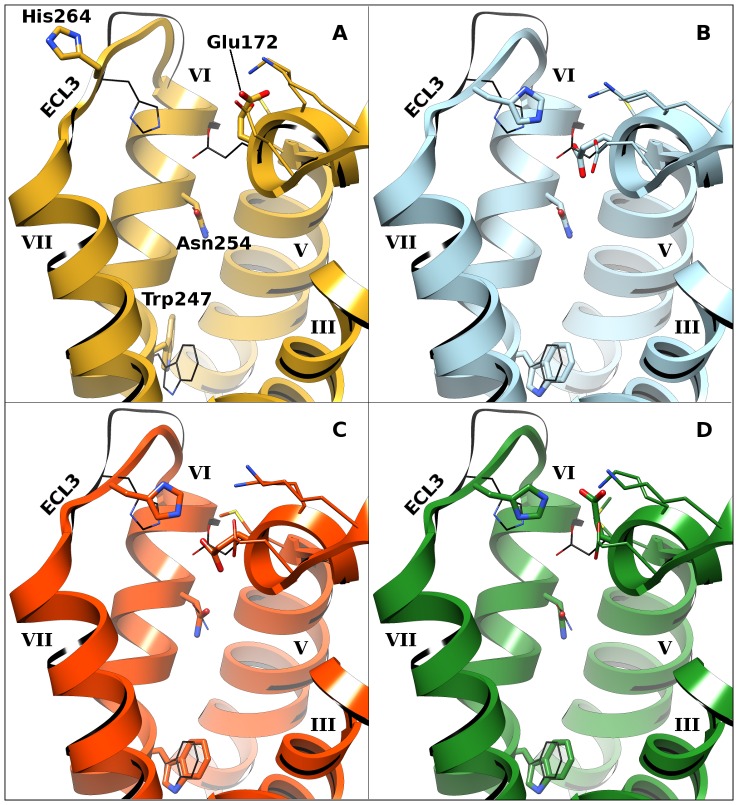
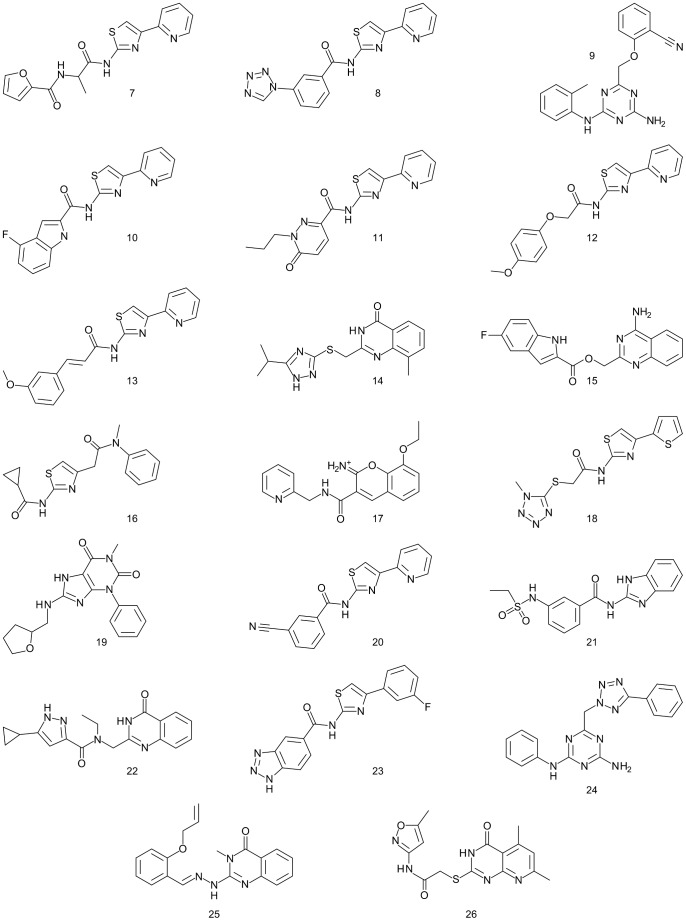
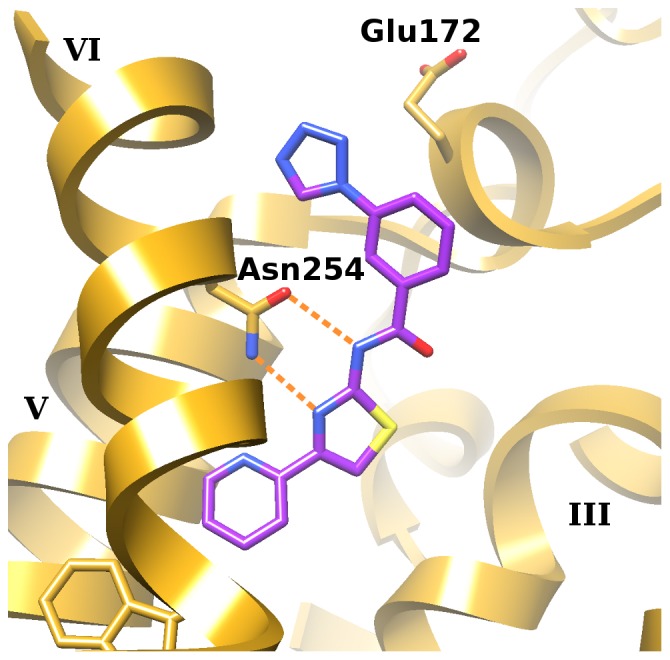
G protein-coupled receptors (GPCRs) are attractive targets for pharmaceutical research. With the recent determination of several GPCR X-ray structures, the applicability of structure-based computational methods for ligand identification, such as docking, has increased. Yet, as only about 1% of GPCRs have a known structure, receptor homology modeling remains necessary. In order to investigate the usability of homology models and the inherent selectivity of a particular model in relation to close homologs, we constructed multiple homology models for the A(1) adenosine receptor (A(1)AR) and docked ∼2.2 M lead-like compounds. High-ranking molecules were tested on the A(1)AR as well as the close homologs A(2A)AR and A(3)AR. While the screen yielded numerous potent and novel ligands (hit rate 21% and highest affinity of 400 nM), it delivered few selective compounds. Moreover, most compounds appeared in the top ranks of only one model. These findings have implications for future screens.
G 蛋白偶联受体(GPCRs)是药物研究的热门靶点。随着最近确定了几种 GPCR 的 X 射线结构,基于结构的计算方法在配体识别方面的适用性,如对接,有所增加。然而,由于只有约 1%的 GPCR 具有已知的结构,受体同源建模仍然是必要的。为了研究同源模型的可用性和特定模型相对于近同源物的固有选择性,我们构建了多个 A(1) 腺苷受体(A(1)AR)的同源模型,并对接了约 220 万个类先导化合物。排名靠前的分子在 A(1)AR 以及近同源物 A(2A)AR 和 A(3)AR 上进行了测试。虽然筛选产生了许多有效且新颖的配体(命中率为 21%,最高亲和力为 400nM),但它提供的选择性化合物却很少。此外,大多数化合物只在一个模型中排名靠前。这些发现对未来的筛选具有启示意义。