UPMC, UMR7238, Génomique Analytique, 15 rue de l'Ecole de Médecine, F-75006 Paris, France.
BMC Bioinformatics. 2012 Aug 8;13:194. doi: 10.1186/1471-2105-13-194.
Searching for similarities in a set of biological data is intrinsically difficult due to possible data points that should not be clustered, or that should group within several clusters. Under these hypotheses, hierarchical agglomerative clustering is not appropriate. Moreover, if the dataset is not known enough, like often is the case, supervised classification is not appropriate either.
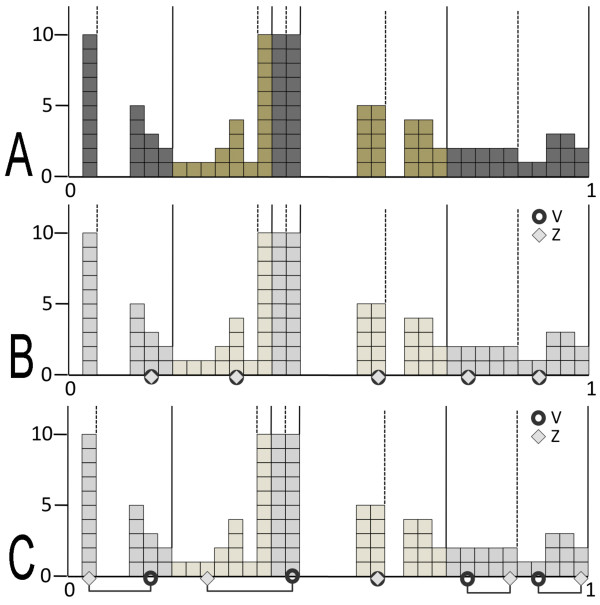
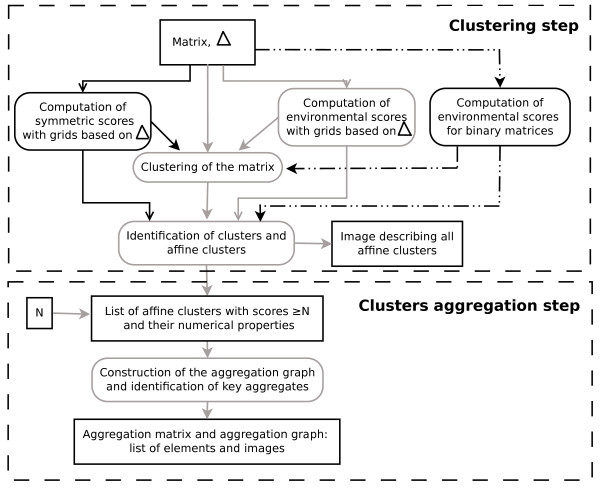
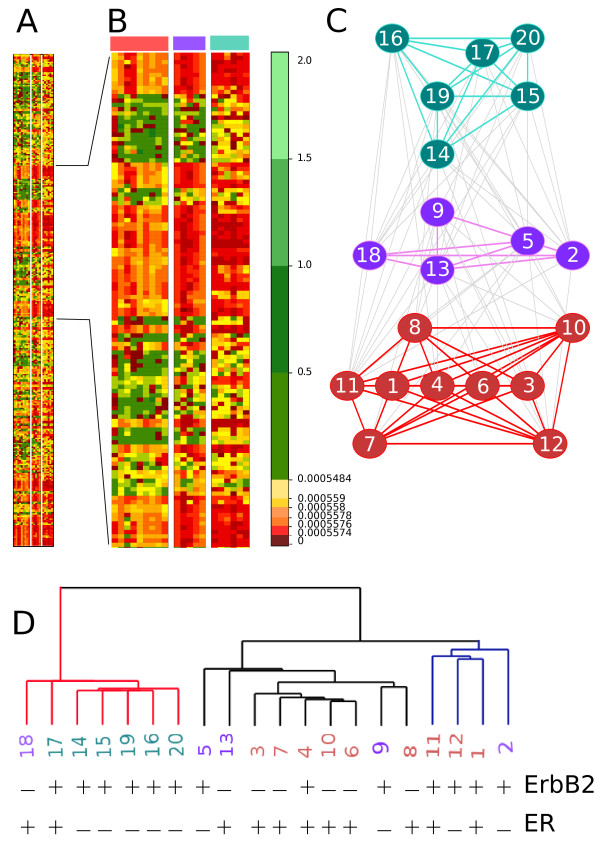
CLAG (for CLusters AGgregation) is an unsupervised non hierarchical clustering algorithm designed to cluster a large variety of biological data and to provide a clustered matrix and numerical values indicating cluster strength. CLAG clusterizes correlation matrices for residues in protein families, gene-expression and miRNA data related to various cancer types, sets of species described by multidimensional vectors of characters, binary matrices. It does not ask to all data points to cluster and it converges yielding the same result at each run. Its simplicity and speed allows it to run on reasonably large datasets.
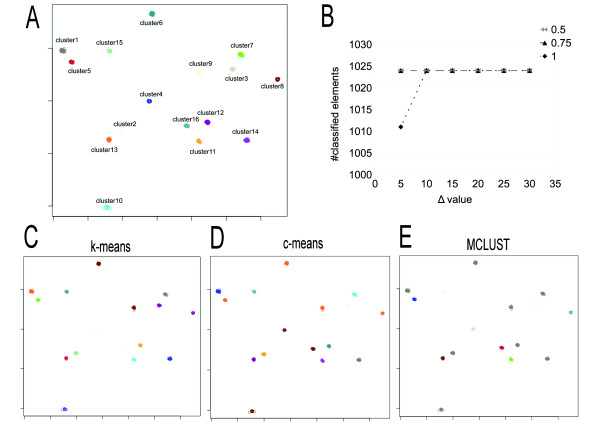
CLAG can be used to investigate the cluster structure present in biological datasets and to identify its underlying graph. It showed to be more informative and accurate than several known clustering methods, as hierarchical agglomerative clustering, k-means, fuzzy c-means, model-based clustering, affinity propagation clustering, and not to suffer of the convergence problem proper to this latter.
由于可能存在不应聚类的数据点,或者应在几个聚类中分组的数据点,因此在一组生物数据中寻找相似性本质上具有难度。在这些假设下,层次凝聚聚类并不合适。此外,如果数据集不够了解,就像通常情况一样,监督分类也不合适。
CLAG(代表聚类聚合)是一种无监督的非层次聚类算法,旨在对各种生物数据进行聚类,并提供聚类矩阵和数值,以指示聚类强度。CLAG 对蛋白质家族中残基的相关矩阵、与各种癌症类型相关的基因表达和 miRNA 数据、由多维字符向量描述的物种集、二进制矩阵进行聚类。它不要求所有数据点进行聚类,并且在每次运行时都会收敛,从而产生相同的结果。其简单性和速度使其能够在相当大的数据集上运行。
CLAG 可用于研究生物数据集的聚类结构,并识别其底层图。与层次凝聚聚类、k-均值、模糊 c-均值、基于模型的聚类、亲和传播聚类等几种已知聚类方法相比,CLAG 更具信息量和准确性,并且不会出现后一种方法特有的收敛问题。