Sorbonne Université, UPMC Univ Paris 06, CNRS, IBPS, UMR 7238, Laboratoire de Biologie Computationnelle et Quantitative (LCQB), 75005 Paris, France.
Nucleic Acids Res. 2020 Jul 2;48(W1):W558-W565. doi: 10.1093/nar/gkaa330.
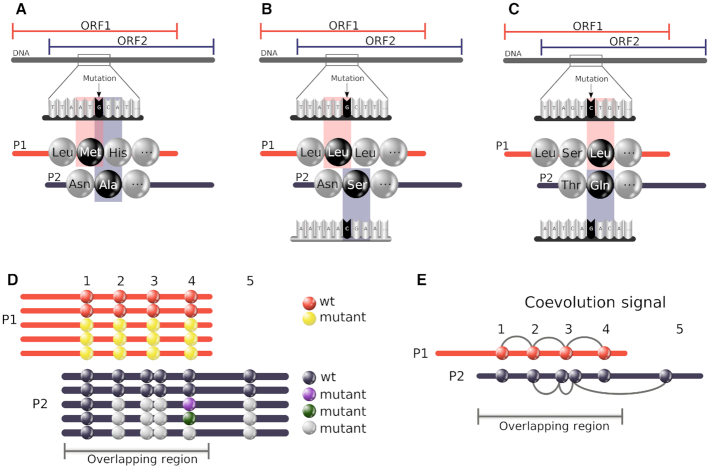
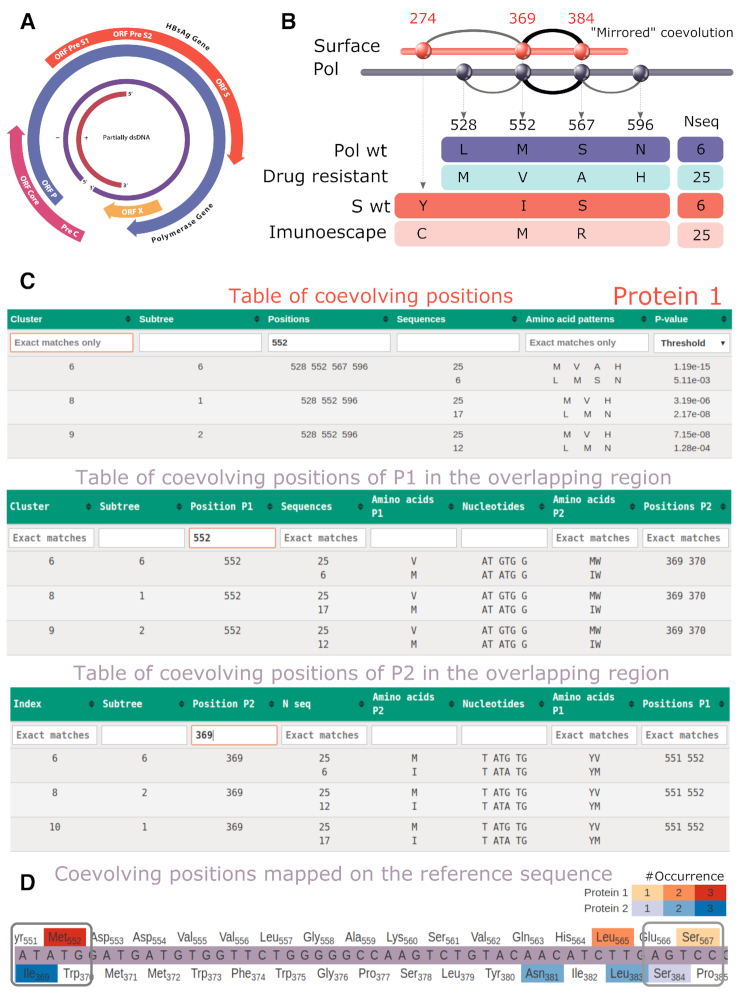
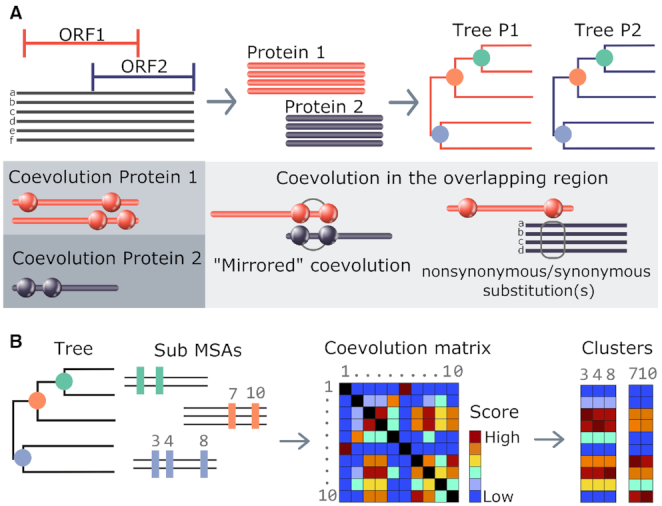
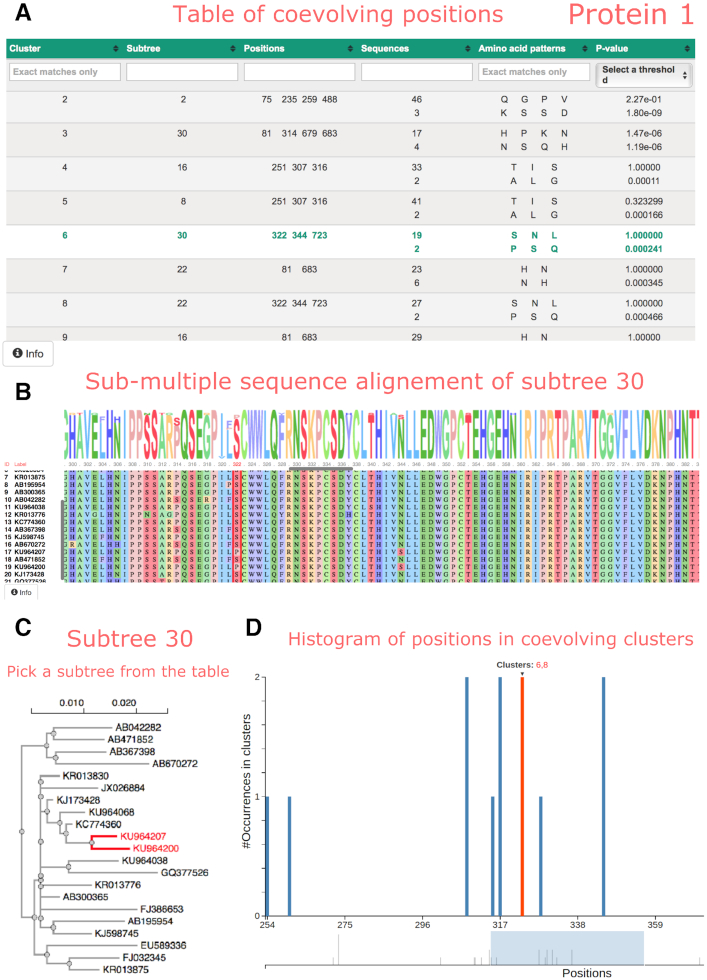
Overlapping genes are commonplace in viruses and play an important role in their function and evolution. For these genes, molecular coevolution may be seen as a mechanism to decrease the evolutionary constraints of amino acid positions in the overlapping regions and to tolerate or compensate unfavorable mutations. Tracing these mutational sites, could help to gain insight on the direct or indirect effect of the mutations in the corresponding overlapping proteins. In the past, coevolution analysis has been used to identify residue pairs and coevolutionary signatures within or between proteins that served as markers of physical interactions and/or functional relationships. Coevolution in OVerlapped sequences by Tree analysis (COVTree) is a web server providing the online analysis of coevolving amino-acid pairs in overlapping genes, where residues might be located inside or outside the overlapping region. COVTree is designed to handle protein families with various characteristics, among which those that typically display a small number of highly conserved sequences. It is based on BIS2, a fast version of the coevolution analysis tool Blocks in Sequences (BIS). COVTree provides a rich and interactive graphical interface to ease biological interpretation of the results and it is openly accessible at http://www.lcqb.upmc.fr/COVTree/.
重叠基因在病毒中很常见,在它们的功能和进化中起着重要作用。对于这些基因,分子共进化可以被视为一种降低重叠区域中氨基酸位置进化约束的机制,并容忍或补偿不利的突变。追踪这些突变位点,可以帮助了解突变在相应重叠蛋白中的直接或间接影响。过去,共进化分析被用于识别蛋白质内部或之间的残基对和共进化特征,这些特征可作为物理相互作用和/或功能关系的标记。通过树分析进行重叠序列中的共进化(COVTree)是一个提供重叠基因中共进化氨基酸对在线分析的网络服务器,其中残基可能位于重叠区域内或外。COVTree 旨在处理具有各种特征的蛋白质家族,其中包括那些通常显示少数高度保守序列的家族。它基于 BIS2,即序列块(BIS)的快速共进化分析工具。COVTree 提供了一个丰富且交互式的图形界面,以方便对结果进行生物学解释,并且可以在 http://www.lcqb.upmc.fr/COVTree/ 上公开访问。