Laboratory for Motor Neuron Disease, RIKEN Brain Science Institute, Wako, Saitama 351-0198, Japan.
J Biol Chem. 2013 Feb 1;288(5):3641-54. doi: 10.1074/jbc.M112.433615. Epub 2012 Dec 12.
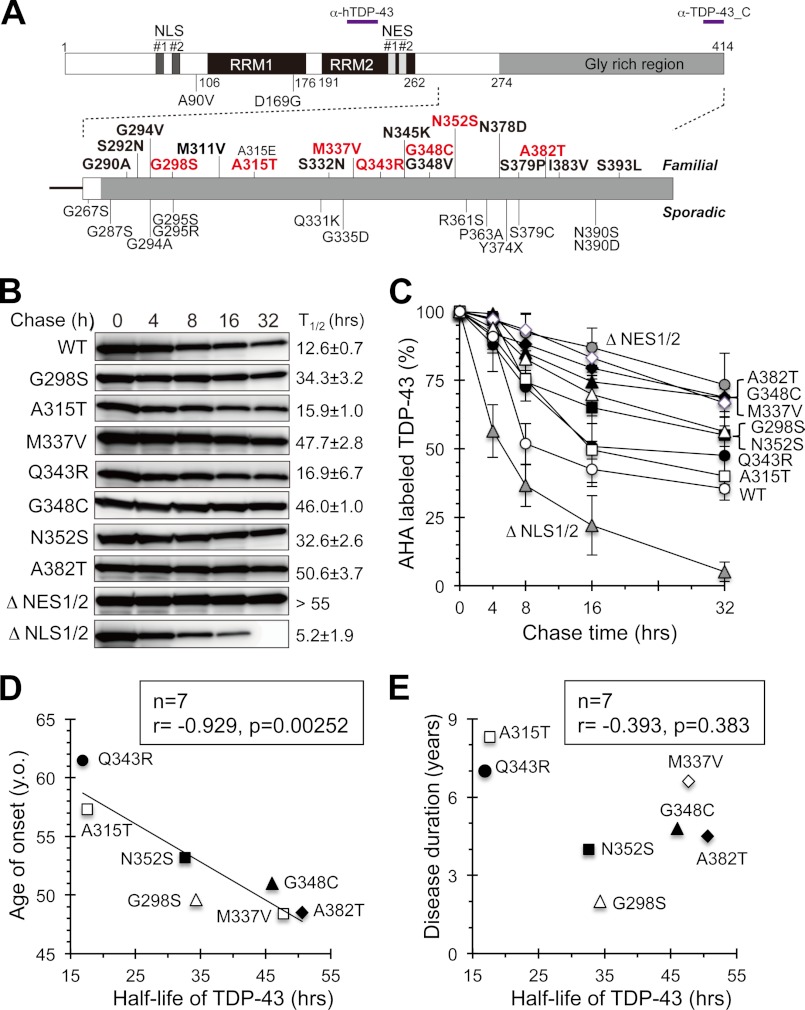
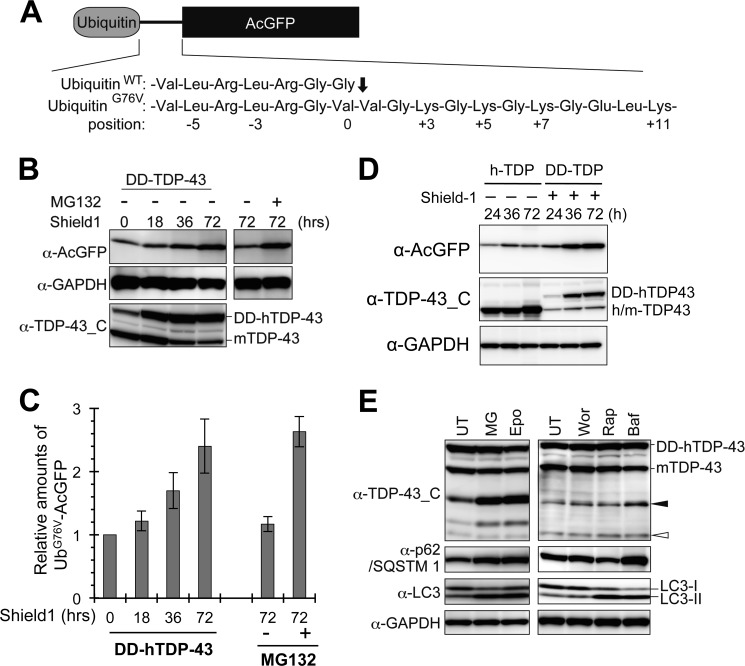
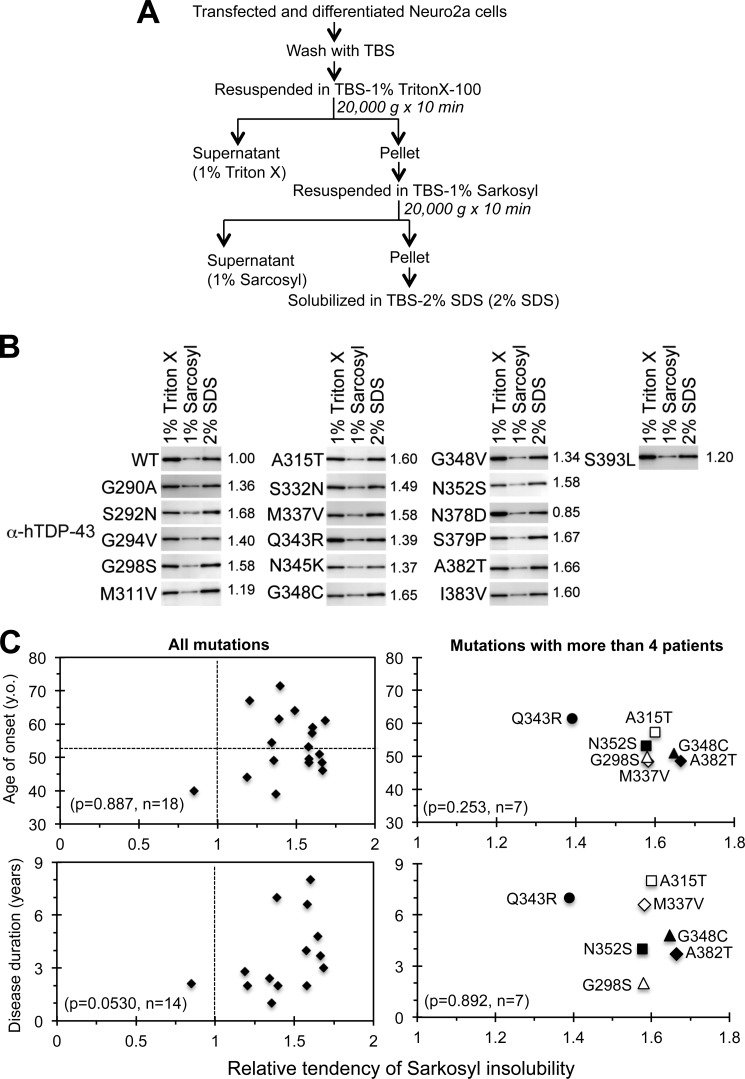
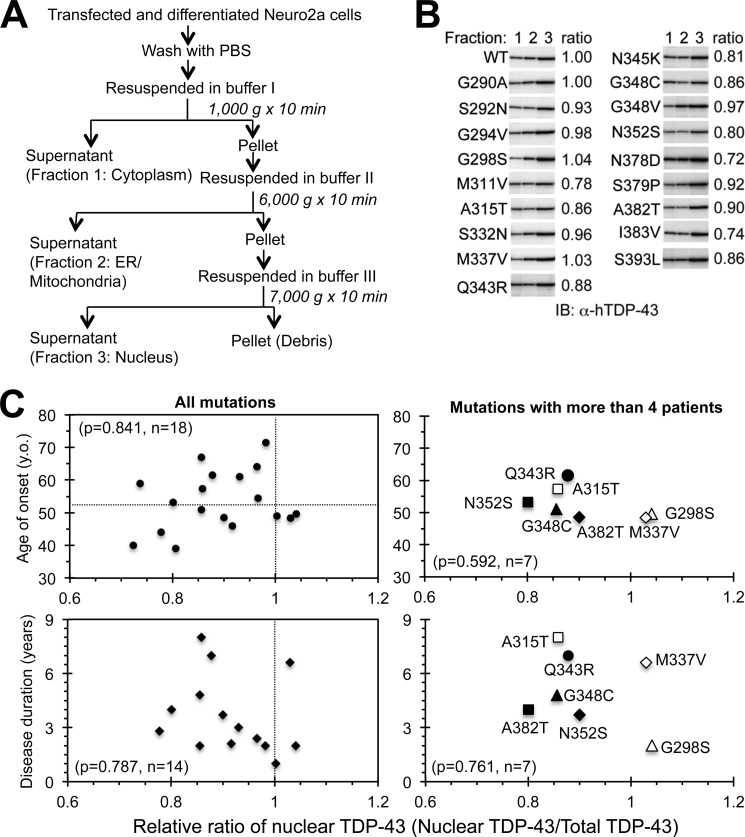
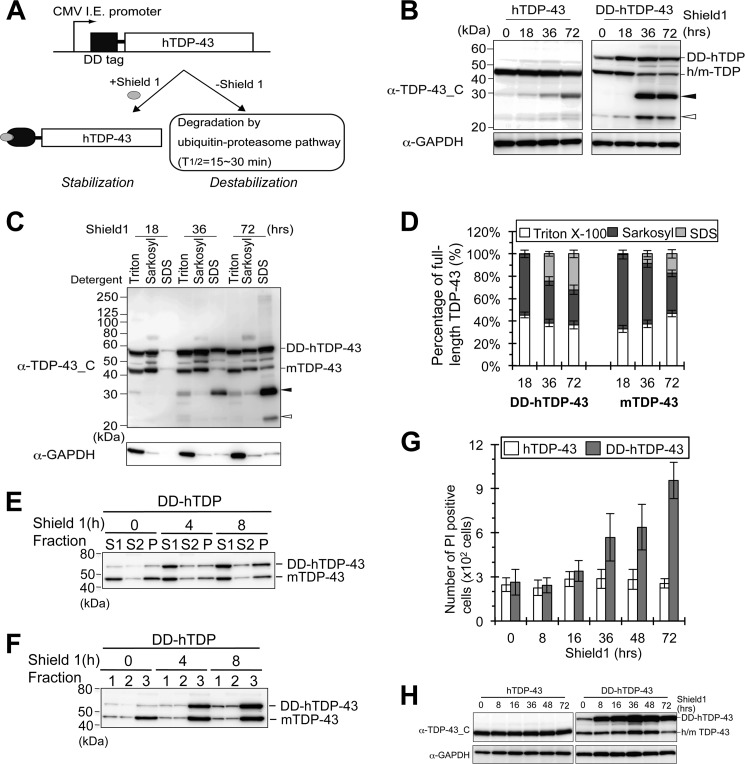
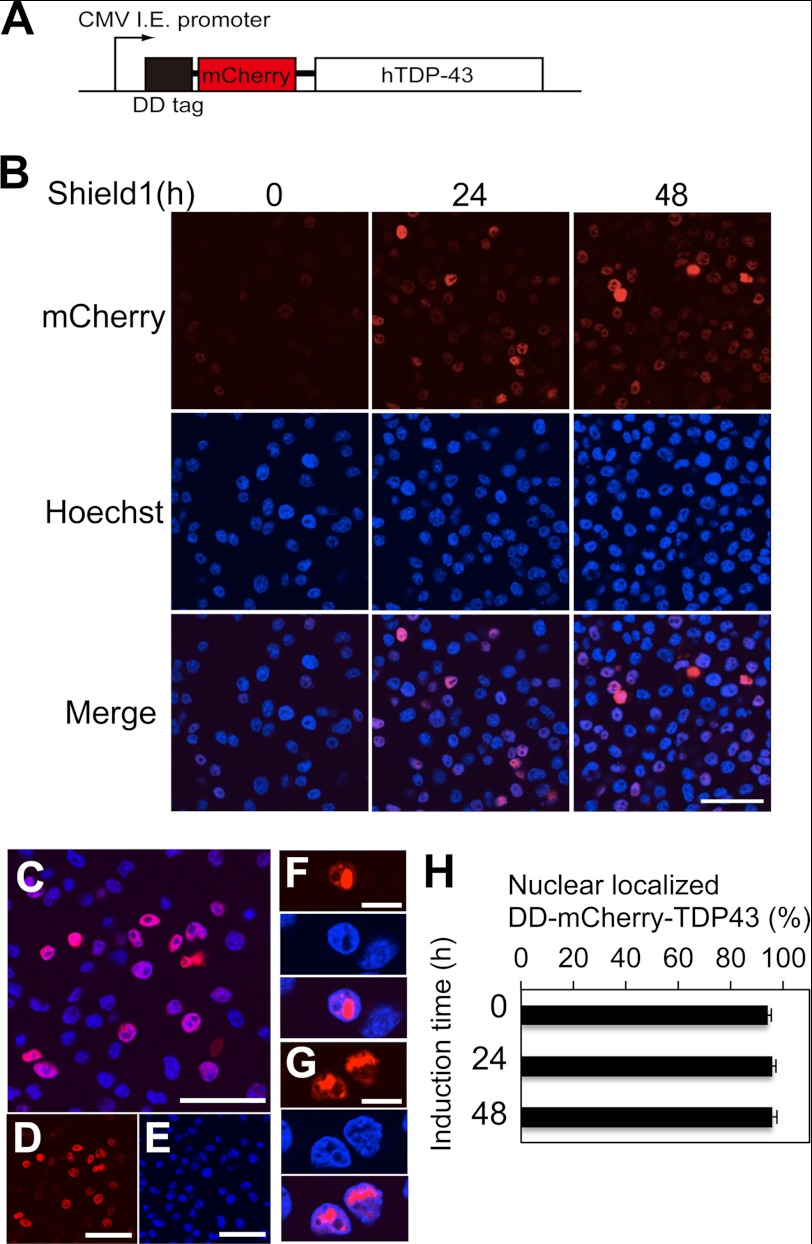
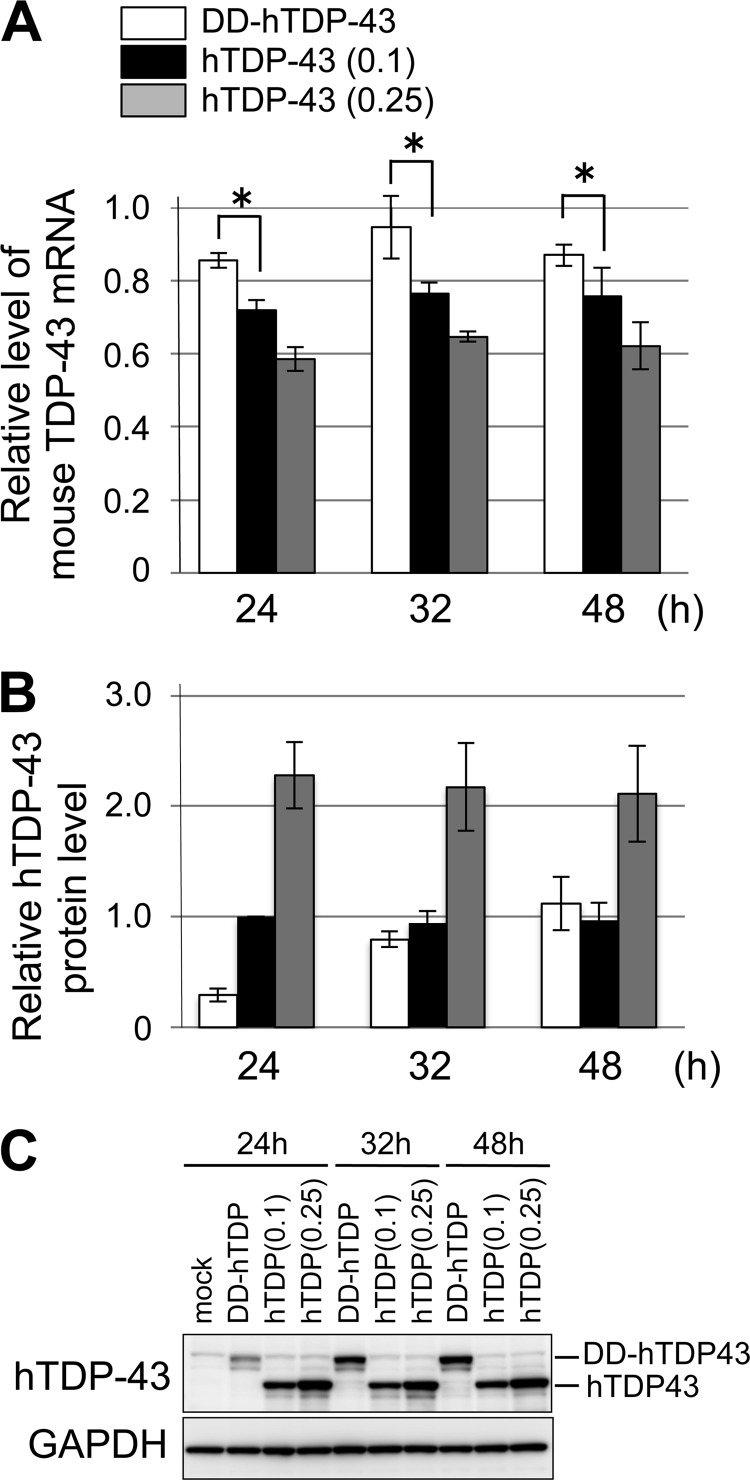
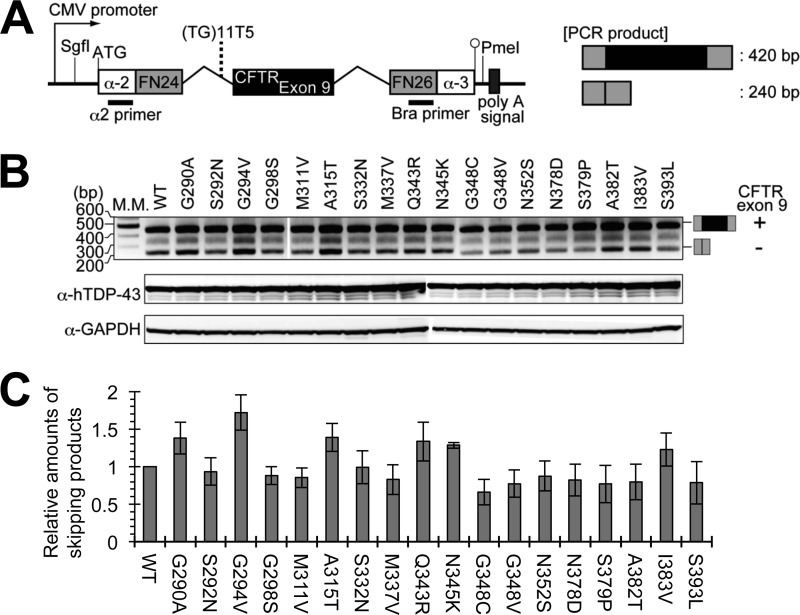
Abnormal protein accumulation is a pathological hallmark of neurodegenerative diseases, including accumulation of TAR DNA-binding protein 43 (TDP-43) in amyotrophic lateral sclerosis (ALS). Dominant mutations in the TDP-43 gene are causative for familial ALS; however, the relationship between mutant protein biochemical phenotypes and disease course and their significance to disease pathomechanism are not known. Here, we found that longer half-lives of mutant proteins correlated with accelerated disease onset. Based on our findings, we established a cell model in which chronic stabilization of wild-type TDP-43 protein provoked cytotoxicity and recapitulated pathogenic protein cleavage and insolubility to the detergent Sarkosyl, TDP-43 properties that have been observed in sporadic ALS lesions. Furthermore, these cells showed proteasomal impairment and dysregulation of their own mRNA levels. These results suggest that chronically increased stability of mutant or wild-type TDP-43 proteins results in a gain of toxicity through abnormal proteostasis.
异常蛋白质积累是神经退行性疾病的病理学标志,包括在肌萎缩侧索硬化症(ALS)中 TAR DNA 结合蛋白 43(TDP-43)的积累。TDP-43 基因的显性突变是家族性 ALS 的原因;然而,突变蛋白生化表型与疾病过程及其对疾病发病机制的意义尚不清楚。在这里,我们发现突变蛋白的半衰期较长与疾病发病加速相关。基于我们的发现,我们建立了一个细胞模型,其中野生型 TDP-43 蛋白的慢性稳定导致了细胞毒性,并重现了致病性蛋白的切割和不溶性到去污剂 Sarkosyl,TDP-43 的特性在散发性 ALS 病变中观察到。此外,这些细胞表现出蛋白酶体损伤和自身 mRNA 水平的失调。这些结果表明,突变型或野生型 TDP-43 蛋白的慢性稳定性增加会通过异常的蛋白质平衡导致毒性增加。