Department of Neuroscience and Pathobiology, Research Institute of Environmental Medicine, Nagoya University, Chikusa-Ku, Nagoya, Aichi, 464-8601, Japan.
Department of Neuroscience and Pathobiology, Graduate School of Medicine, Nagoya University, Nagoya, Aichi, 466-8550, Japan.
Acta Neuropathol Commun. 2024 Nov 27;12(1):184. doi: 10.1186/s40478-024-01893-x.
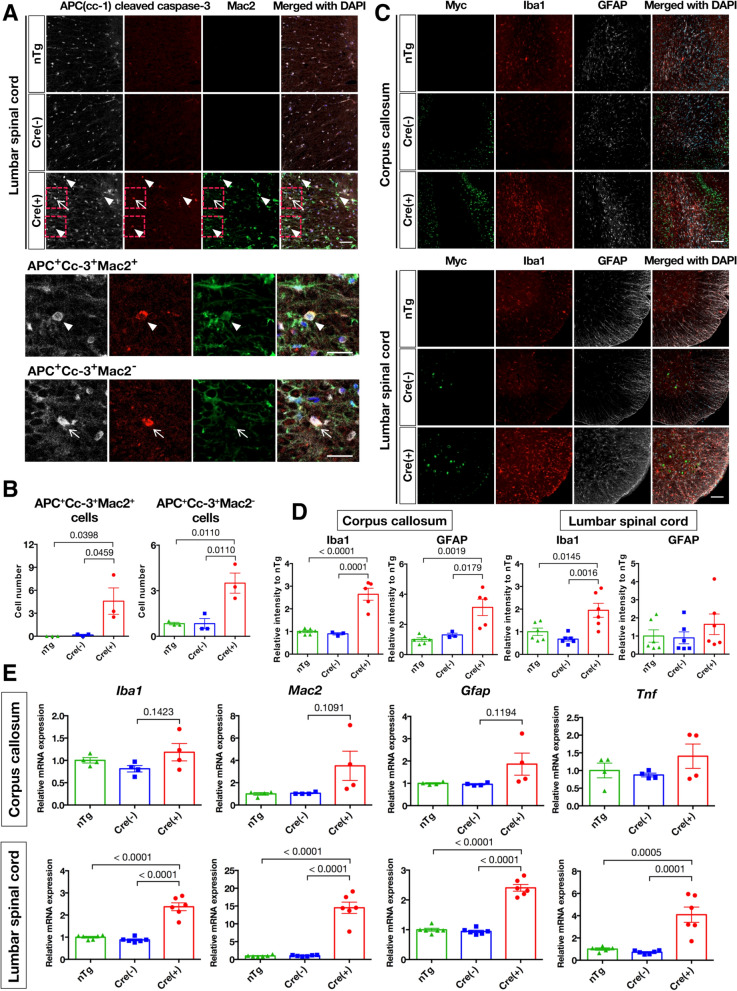
Nuclear clearance and cytoplasmic aggregation of TAR DNA-binding protein of 43 kDa (TDP-43) are pathological hallmarks of amyotrophic lateral sclerosis (ALS) and its pathogenic mechanism is mediated by both loss-of-function and gain-of-toxicity of TDP-43. However, the role of TDP-43 gain-of-toxicity in oligodendrocytes remains unclear. To investigate the impact of excess TDP-43 on oligodendrocytes, we established transgenic mice overexpressing the ALS-linked mutant TDP-43 in oligodendrocytes through crossbreeding with Mbp-Cre mice. Two-step crossbreeding of floxed TDP-43 and Mbp-Cre mice resulted in the heterozygous low-level systemic expression of TDP-43 with (Cre-positive) or without (Cre-negative) oligodendrocyte-specific overexpression of TDP-43. Although Cre-negative mice also exhibit subtle motor dysfunction, TDP-43 overexpression in oligodendrocytes aggravated clasping signs and gait disturbance accompanied by myelin pallor in the corpus callosum and white matter of the lumbar spinal cord in Cre-positive mice. RNA sequencing analysis of oligodendrocyte lineage cells isolated from whole brains of 12-month-old transgenic mice revealed downregulation of myelinating oligodendrocyte marker genes and cholesterol-related genes crucial for myelination, along with marked upregulation of apoptotic pathway genes. Immunofluorescence staining showed cleaved caspase 3-positive apoptotic oligodendrocytes surrounded by activated microglia and astrocytes in aged transgenic mice. Collectively, our findings demonstrate that an excess amount of ALS-linked mutant TDP-43 expression in oligodendrocytes exacerbates motor dysfunction in mice, likely through oligodendrocyte dysfunction and neuroinflammation. Therefore, targeting oligodendrocyte protection, particularly through ameliorating TDP-43 pathology, could represent a potential therapeutic approach for ALS.
TDP-43 蛋白的核清除和细胞质聚集是肌萎缩侧索硬化症(ALS)的病理标志,其致病机制既涉及 TDP-43 的功能丧失,也涉及毒性获得。然而,TDP-43 毒性获得在少突胶质细胞中的作用仍不清楚。为了研究过量 TDP-43 对少突胶质细胞的影响,我们通过与 Mbp-Cre 小鼠杂交,在少突胶质细胞中建立了表达 ALS 相关突变 TDP-43 的转基因小鼠。通过两步杂交 floxed TDP-43 和 Mbp-Cre 小鼠,导致 TDP-43 在系统中的低水平杂合表达(Cre 阳性)或缺乏(Cre 阴性)少突胶质细胞特异性过表达。尽管 Cre 阴性小鼠也表现出轻微的运动功能障碍,但 Cre 阳性小鼠中 TDP-43 在少突胶质细胞中的过表达加剧了扣状征和步态障碍,同时伴有胼胝体和腰椎脊髓白质的髓鞘苍白。对来自 12 个月大转基因小鼠全脑分离的少突胶质细胞谱系细胞的 RNA 测序分析显示,髓鞘形成少突胶质细胞标记基因和髓鞘形成所必需的胆固醇相关基因下调,同时凋亡途径基因显著上调。免疫荧光染色显示,在老年转基因小鼠中,被激活的小胶质细胞和星形胶质细胞包围的凋亡少突胶质细胞中存在 cleaved caspase 3 阳性凋亡少突胶质细胞。总之,我们的研究结果表明,过量的 ALS 相关突变 TDP-43 在少突胶质细胞中的表达加剧了小鼠的运动功能障碍,可能是通过少突胶质细胞功能障碍和神经炎症。因此,针对少突胶质细胞的保护,特别是通过改善 TDP-43 病理学,可能代表 ALS 的一种潜在治疗方法。