Dubowitz Neuromuscular Centre, UCL Institute of Child Health, University College London, 30 Guilford Street, London WC1N 1EH, UK.
Brain. 2013 Jan;136(Pt 1):269-81. doi: 10.1093/brain/aws312. Epub 2013 Jan 3.
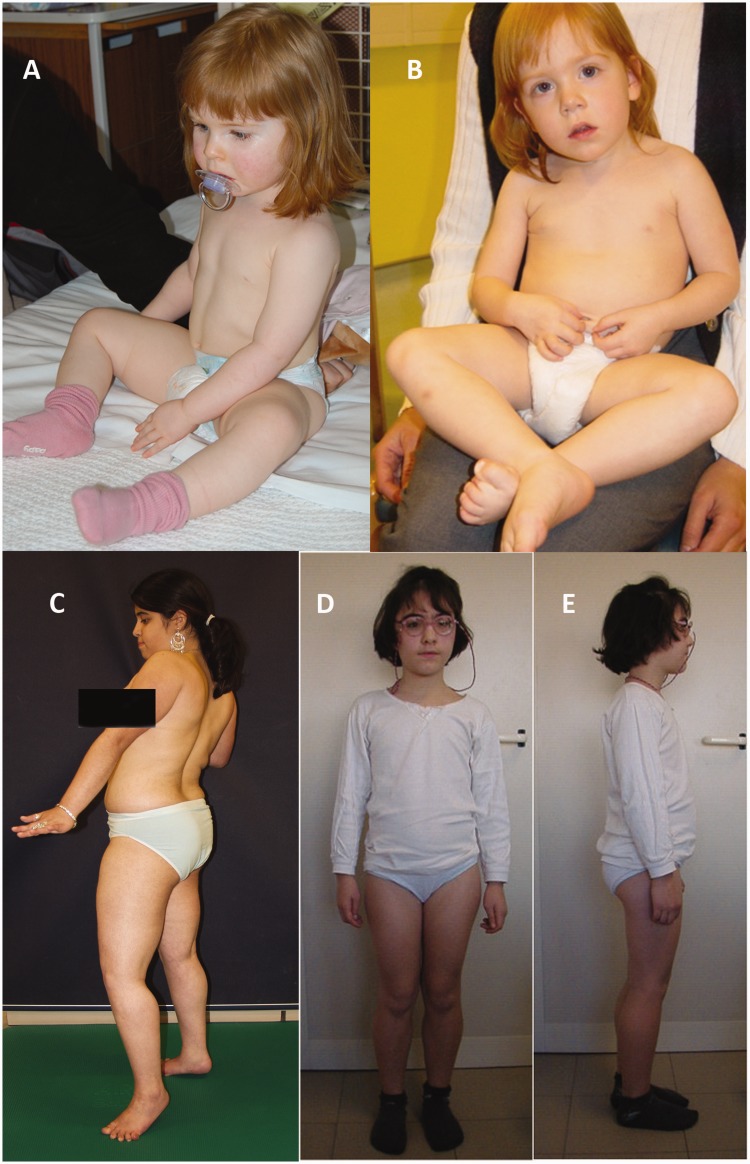
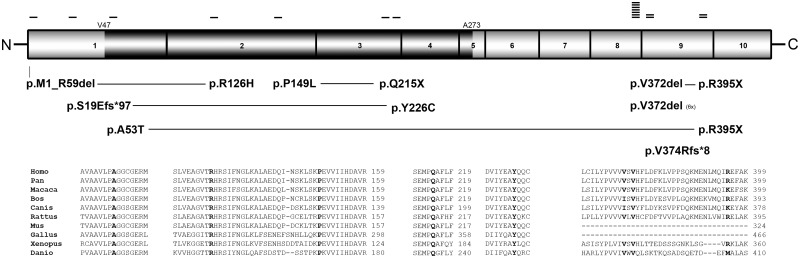
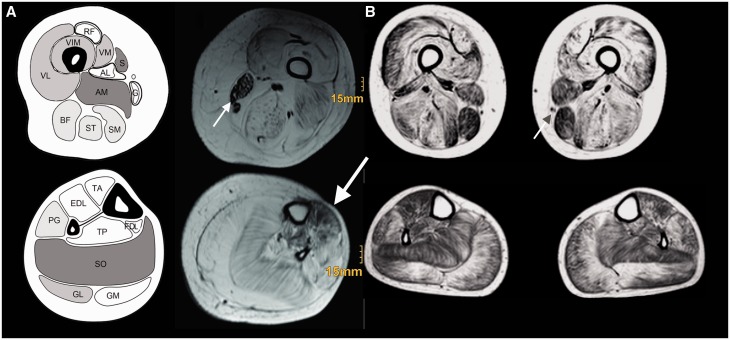
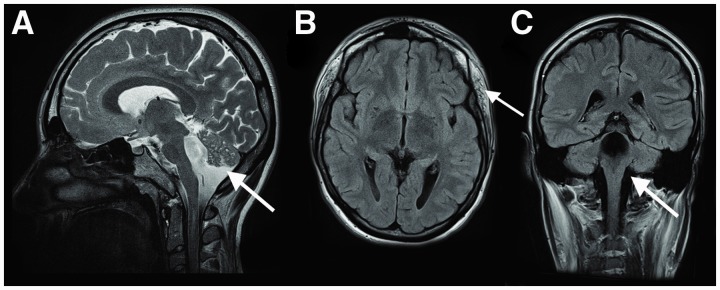
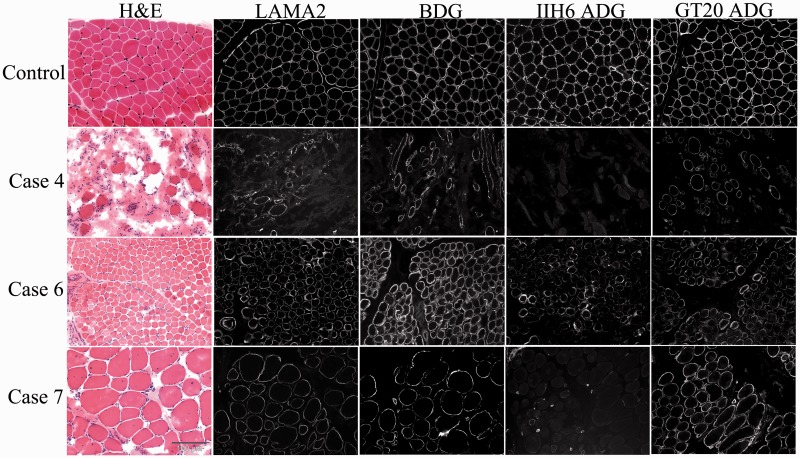
Dystroglycanopathies are a clinically and genetically diverse group of recessively inherited conditions ranging from the most severe of the congenital muscular dystrophies, Walker-Warburg syndrome, to mild forms of adult-onset limb-girdle muscular dystrophy. Their hallmark is a reduction in the functional glycosylation of α-dystroglycan, which can be detected in muscle biopsies. An important part of this glycosylation is a unique O-mannosylation, essential for the interaction of α-dystroglycan with extracellular matrix proteins such as laminin-α2. Mutations in eight genes coding for proteins in the glycosylation pathway are responsible for ∼50% of dystroglycanopathy cases. Despite multiple efforts using traditional positional cloning, the causative genes for unsolved dystroglycanopathy cases have escaped discovery for several years. In a recent collaborative study, we discovered that loss-of-function recessive mutations in a novel gene, called isoprenoid synthase domain containing (ISPD), are a relatively common cause of Walker-Warburg syndrome. In this article, we report the involvement of the ISPD gene in milder dystroglycanopathy phenotypes ranging from congenital muscular dystrophy to limb-girdle muscular dystrophy and identified allelic ISPD variants in nine cases belonging to seven families. In two ambulant cases, there was evidence of structural brain involvement, whereas in seven, the clinical manifestation was restricted to a dystrophic skeletal muscle phenotype. Although the function of ISPD in mammals is not yet known, mutations in this gene clearly lead to a reduction in the functional glycosylation of α-dystroglycan, which not only causes the severe Walker-Warburg syndrome but is also a common cause of the milder forms of dystroglycanopathy.
肌营养不良糖蛋白病是一组临床表现和遗传方式均具有高度异质性的隐性遗传性疾病,范围广泛,从轻型的先天性肌营养不良(沃勒变性肌病)到成年起病的肢带型肌营养不良症等。其特征为α- 肌营养不良糖蛋白的功能糖基化减少,可在肌肉活检中检测到。这种糖基化的一个重要部分是独特的 O-甘露糖基化,这对于 α- 肌营养不良糖蛋白与细胞外基质蛋白如层粘连蛋白-α2 的相互作用至关重要。糖基化途径中 8 个编码蛋白的基因突变导致约 50%的肌营养不良糖蛋白病病例。尽管使用传统的定位克隆进行了多次尝试,但多年来未能发现未解决的肌营养不良糖蛋白病病例的致病基因。在最近的一项合作研究中,我们发现新型基因异戊烯基转移酶结构域包含(ISPD)的失功能隐性突变是沃勒变性肌病的一个相对常见的原因。在本文中,我们报告了 ISPD 基因在从先天性肌营养不良症到肢带型肌营养不良症等较轻的肌营养不良糖蛋白病表型中的参与,并在属于七个家庭的九例中鉴定出等位基因 ISPD 变体。在两名可走动的病例中,有证据表明存在结构性脑受累,而在七名病例中,临床表现仅限于进行性肌肉营养不良的骨骼表型。尽管哺乳动物中 ISPD 的功能尚不清楚,但该基因的突变显然导致α- 肌营养不良糖蛋白的功能糖基化减少,这不仅导致严重的沃勒变性肌病,也是较轻形式的肌营养不良糖蛋白病的常见原因。