Global and Tropical Health Division, Menzies School of Health Research, Charles Darwin University, Darwin, Australia.
PLoS One. 2013;8(1):e53160. doi: 10.1371/journal.pone.0053160. Epub 2013 Jan 4.
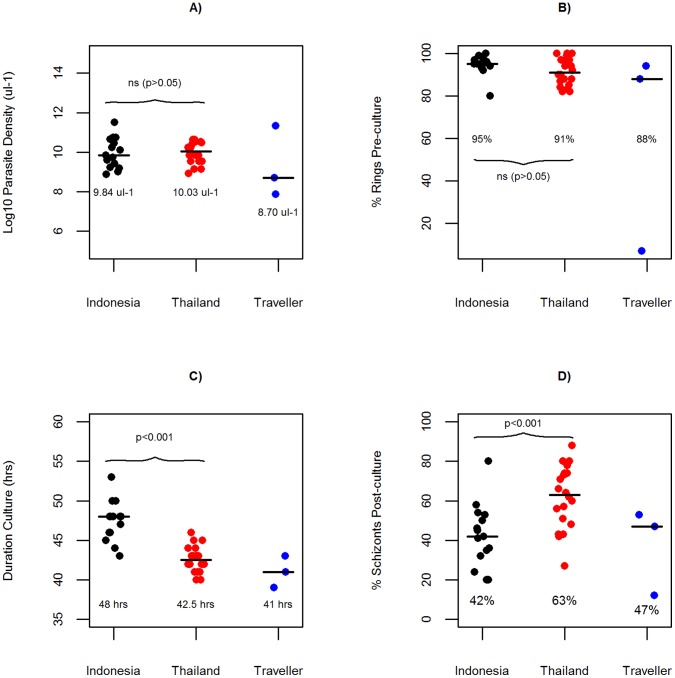
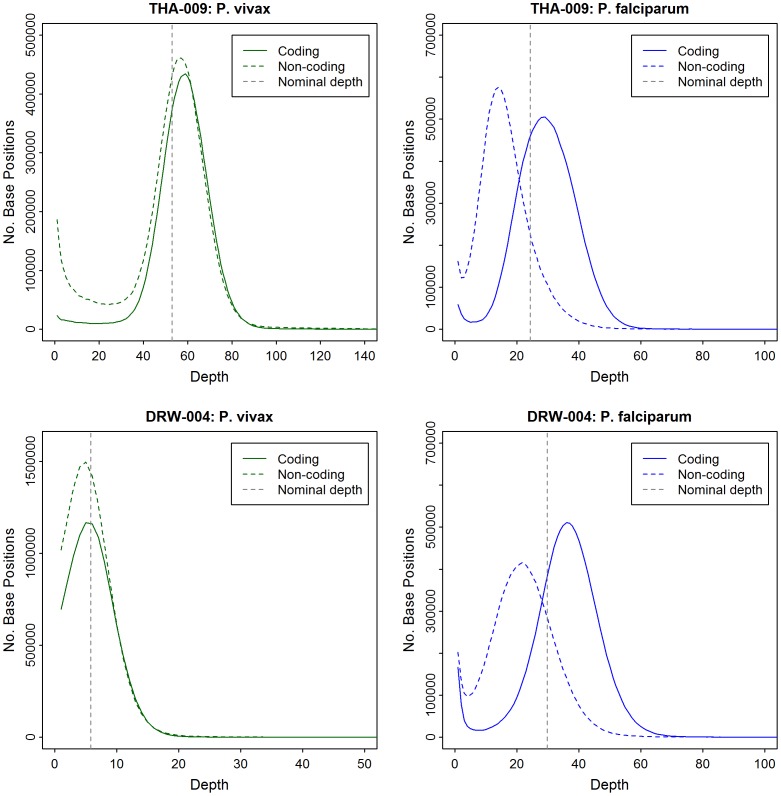
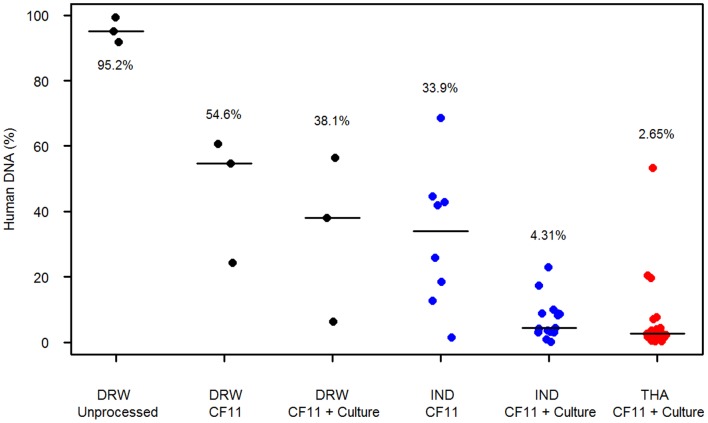
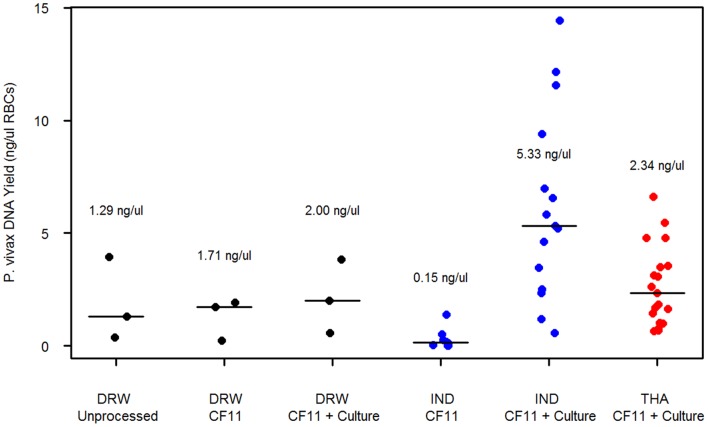
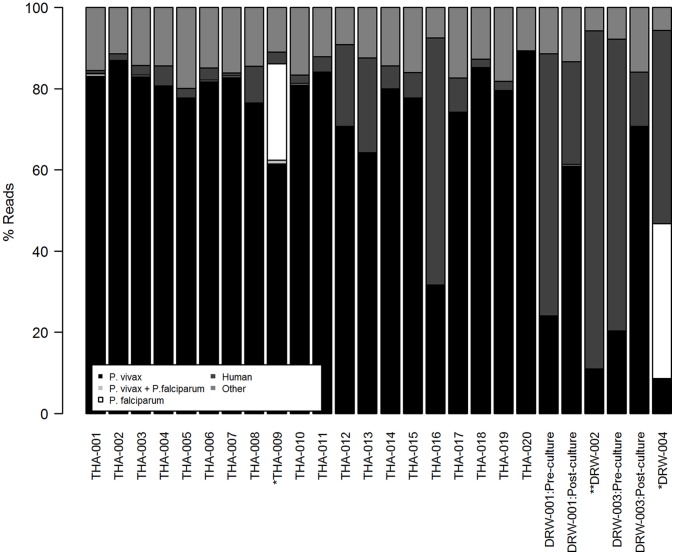
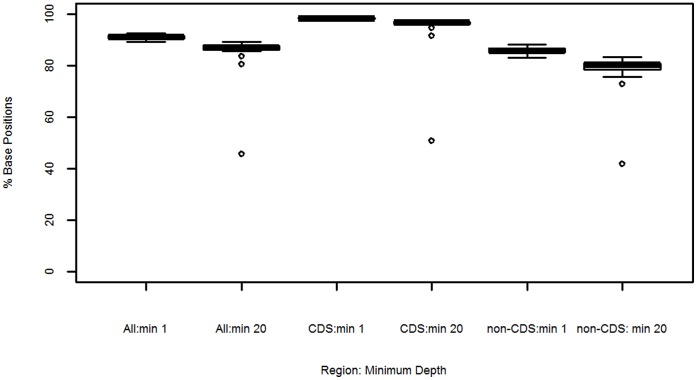
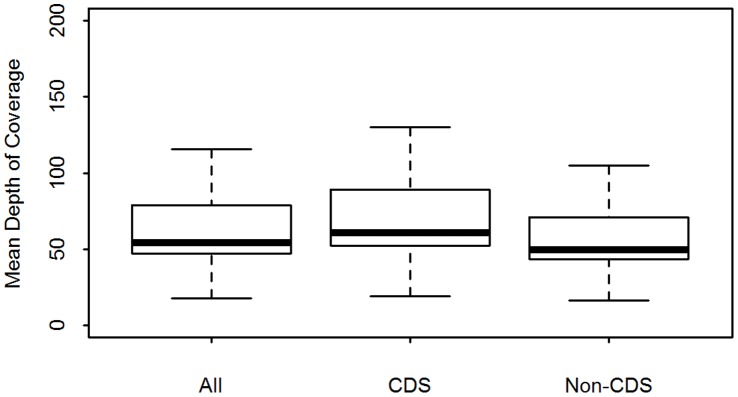
Whole genome sequencing (WGS) of Plasmodium vivax is problematic due to the reliance on clinical isolates which are generally low in parasitaemia and sample volume. Furthermore, clinical isolates contain a significant contaminating background of host DNA which confounds efforts to map short read sequence of the target P. vivax DNA. Here, we discuss a methodology to significantly improve the success of P. vivax WGS on natural (non-adapted) patient isolates. Using 37 patient isolates from Indonesia, Thailand, and travellers, we assessed the application of CF11-based white blood cell filtration alone and in combination with short term ex vivo schizont maturation. Although CF11 filtration reduced human DNA contamination in 8 Indonesian isolates tested, additional short-term culture increased the P. vivax DNA yield from a median of 0.15 to 6.2 ng µl(-1) packed red blood cells (pRBCs) (p = 0.001) and reduced the human DNA percentage from a median of 33.9% to 6.22% (p = 0.008). Furthermore, post-CF11 and culture samples from Thailand gave a median P. vivax DNA yield of 2.34 ng µl(-1) pRBCs, and 2.65% human DNA. In 22 P. vivax patient isolates prepared with the 2-step method, we demonstrate high depth (median 654X coverage) and breadth (≥89%) of coverage on the Illumina GAII and HiSeq platforms. In contrast to the A+T-rich P. falciparum genome, negligible bias was observed in coverage depth between coding and non-coding regions of the P. vivax genome. This uniform coverage will greatly facilitate the detection of SNPs and copy number variants across the genome, enabling unbiased exploration of the natural diversity in P. vivax populations.
全基因组测序(WGS)由于依赖于临床分离株而存在问题,这些分离株通常寄生虫血症和样本量较低。此外,临床分离株中含有大量宿主 DNA 的污染背景,这使得对目标 P. vivax DNA 的短读序列进行映射变得复杂。在这里,我们讨论了一种大大提高天然(非适应)患者分离株 P. vivax WGS 成功率的方法。使用来自印度尼西亚、泰国和旅行者的 37 个患者分离株,我们评估了单独使用基于 CF11 的白细胞过滤以及与短期体外裂殖体成熟相结合的应用。尽管 CF11 过滤减少了 8 个测试的印度尼西亚分离株中的人类 DNA 污染,但额外的短期培养将 P. vivax DNA 产量从中位数的 0.15 增加到 6.2 ng µl(-1) 浓缩红细胞(pRBC)(p = 0.001),并将人类 DNA 的百分比从中位数的 33.9%降低到 6.22%(p = 0.008)。此外,来自泰国的 CF11 后和培养样本的 P. vivax DNA 产量中位数为 2.34 ng µl(-1) pRBC,人类 DNA 为 2.65%。在使用 2 步法制备的 22 个 P. vivax 患者分离株中,我们在 Illumina GAII 和 HiSeq 平台上证明了高深度(中位数 654X 覆盖率)和广度(≥89%)的覆盖率。与 A+T 丰富的 P. falciparum 基因组相比,在 P. vivax 基因组的编码和非编码区域之间,观察到的覆盖率深度几乎没有偏差。这种均匀的覆盖范围将极大地促进在整个基因组中 SNP 和拷贝数变异的检测,从而能够对 P. vivax 种群的自然多样性进行无偏探索。