National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA.
Genome Biol Evol. 2013;5(1):233-42. doi: 10.1093/gbe/evt002.
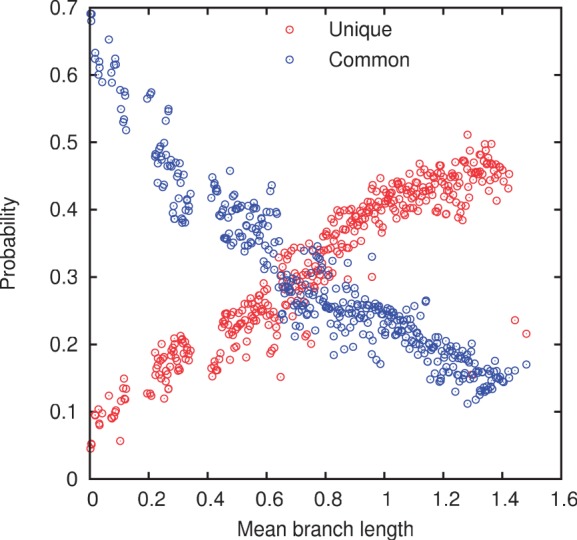
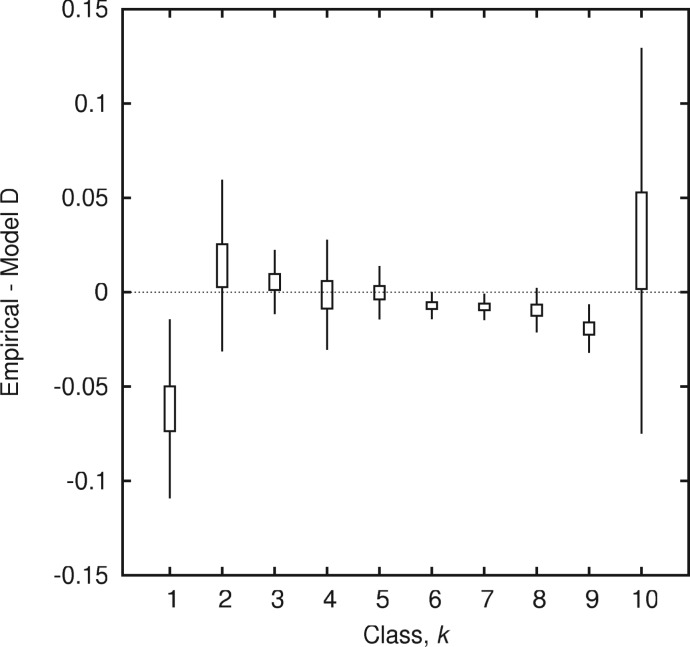
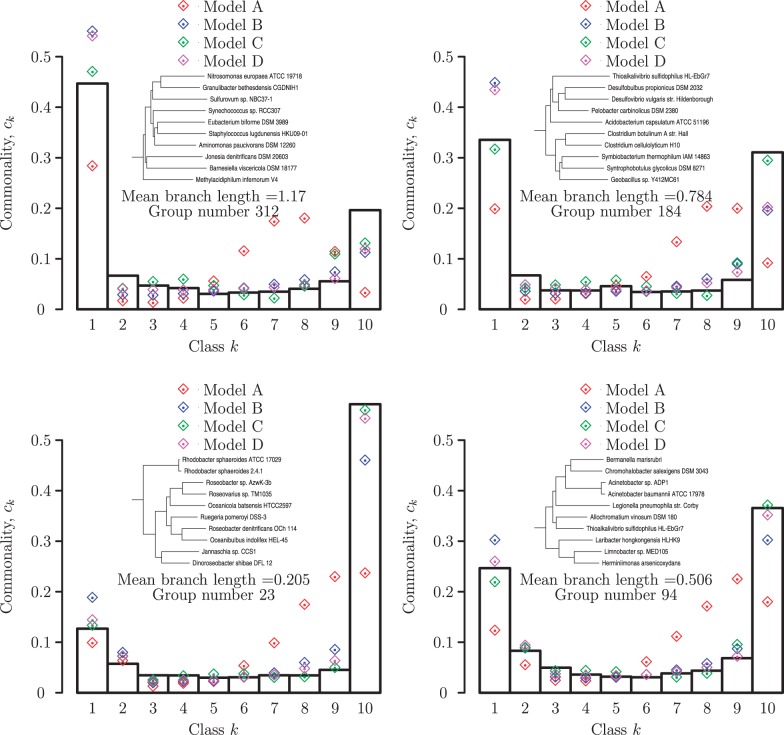
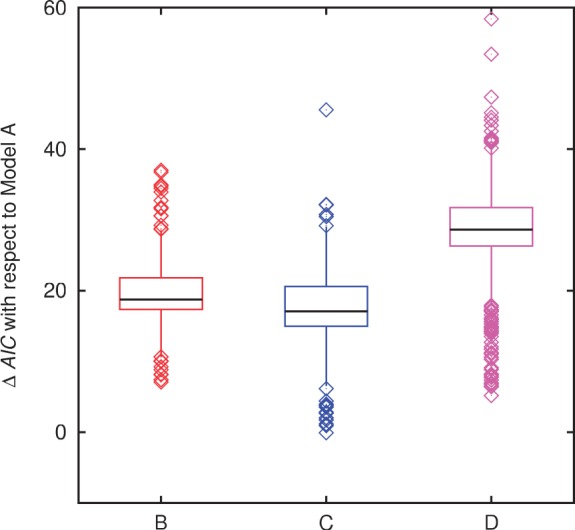
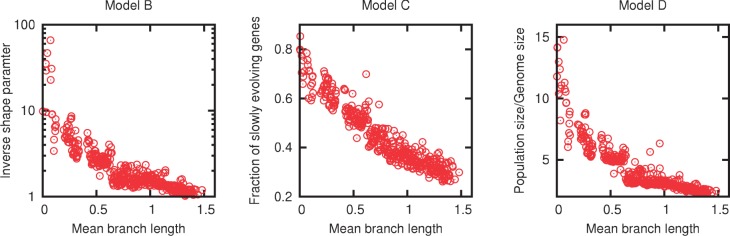
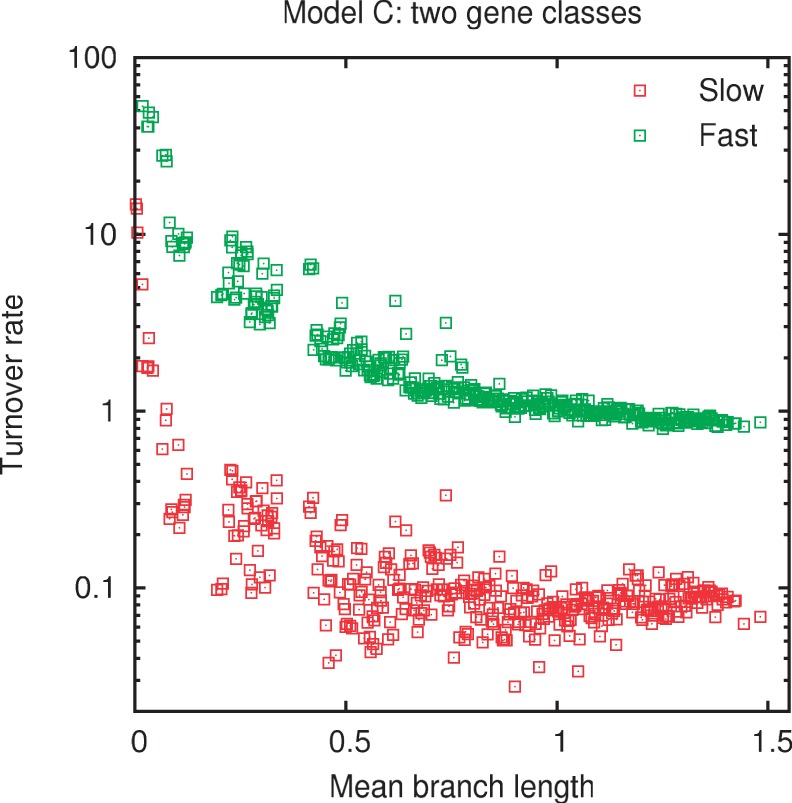
Evolution of prokaryotes involves extensive loss and gain of genes, which lead to substantial differences in the gene repertoires even among closely related organisms. Through a wide range of phylogenetic depths, gene frequency distributions in prokaryotic pangenomes bear a characteristic, asymmetrical U-shape, with a core of (nearly) universal genes, a "shell" of moderately common genes, and a "cloud" of rare genes. We employ mathematical modeling to investigate evolutionary processes that might underlie this universal pattern. Gene frequency distributions for almost 400 groups of 10 bacterial or archaeal species each over a broad range of evolutionary distances were fit to steady-state, infinite allele models based on the distribution of gene replacement rates and the phylogenetic tree relating the species in each group. The fits of the theoretical frequency distributions to the empirical ones yield model parameters and estimates of the goodness of fit. Using the Akaike Information Criterion, we show that the neutral model of genome evolution, with the same replacement rate for all genes, can be confidently rejected. Of the three tested models with purifying selection, the one in which the distribution of replacement rates is derived from a stochastic population model with additive per-gene fitness yields the best fits to the data. The selection strength estimated from the fits declines with evolutionary divergence while staying well outside the neutral regime. These findings indicate that, unlike some other universal distributions of genomic variables, for example, the distribution of paralogous gene family membership, the gene frequency distribution is substantially affected by selection.
原核生物的进化涉及到基因的广泛丢失和获得,这导致即使在密切相关的生物之间,基因谱也存在很大的差异。通过广泛的系统发育深度,原核泛基因组中的基因频率分布呈现出一种特征性的、不对称的 U 形,其中有一个(几乎)普遍存在的基因核心,一个中等常见基因的“外壳”,以及一个稀有基因的“云”。我们运用数学建模来研究可能构成这种普遍模式的进化过程。我们拟合了几乎 400 组每组包含 10 个细菌或古菌的物种的基因频率分布,这些物种在广泛的进化距离上都有分布,拟合基于基因替换率的分布和将每组物种联系起来的系统发育树。理论频率分布与经验频率分布的拟合产生了模型参数和拟合优度的估计。使用赤池信息量准则,我们表明,所有基因具有相同替换率的基因组进化中性模型可以被自信地拒绝。在三种经过测试的具有纯化选择的模型中,基于具有加性基因适应度的随机种群模型推导出的替换率分布的模型,对数据的拟合效果最好。从拟合中估计的选择强度随进化分歧而下降,但仍远在中性范围内。这些发现表明,与其他一些普遍的基因组变量分布不同,例如,旁系同源基因家族成员的分布,基因频率分布受到选择的显著影响。