Department of Cancer Research and Molecular Medicine, Norwegian University of Science and Technology, Trondheim, Norway.
BMC Bioinformatics. 2013 Jan 16;14:9. doi: 10.1186/1471-2105-14-9.
Traditional methods for computational motif discovery often suffer from poor performance. In particular, methods that search for sequence matches to known binding motifs tend to predict many non-functional binding sites because they fail to take into consideration the biological state of the cell. In recent years, genome-wide studies have generated a lot of data that has the potential to improve our ability to identify functional motifs and binding sites, such as information about chromatin accessibility and epigenetic states in different cell types. However, it is not always trivial to make use of this data in combination with existing motif discovery tools, especially for researchers who are not skilled in bioinformatics programming.
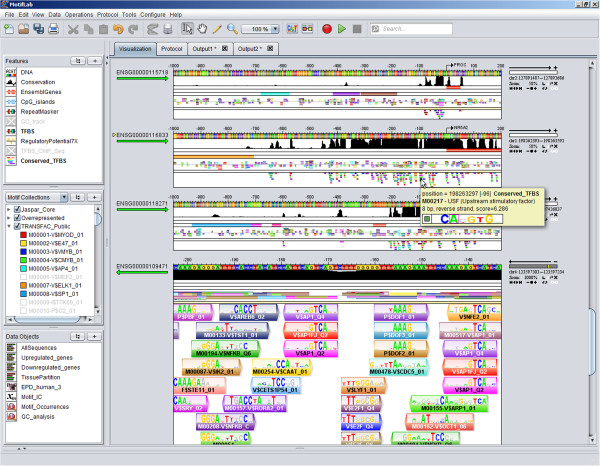
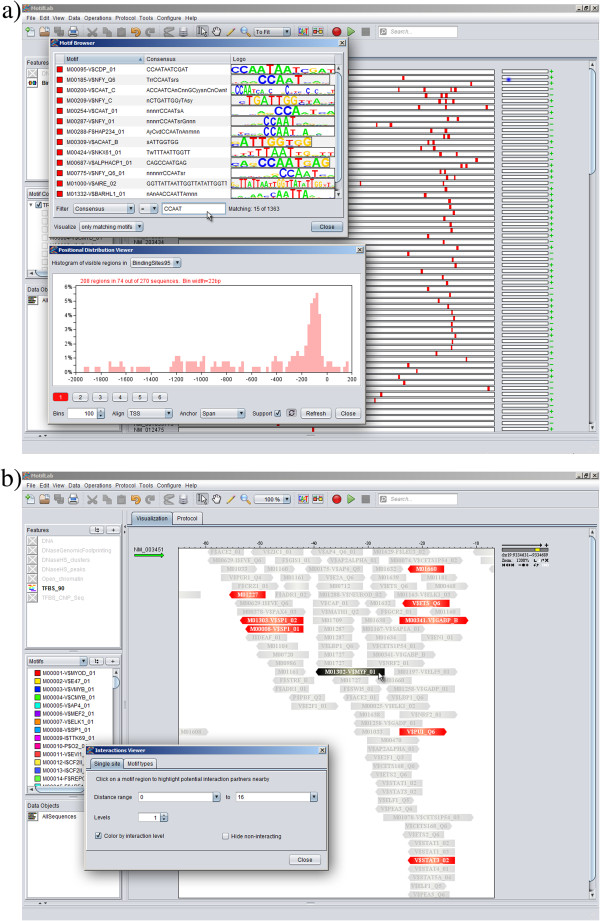
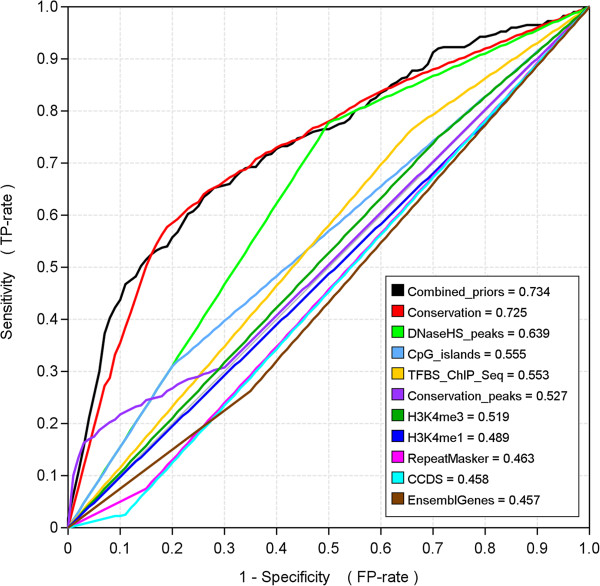
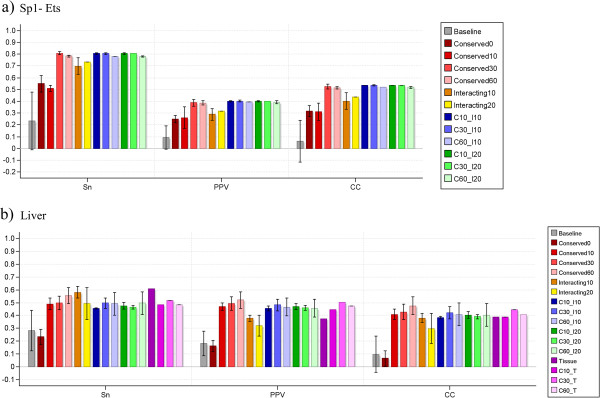
Here we present MotifLab, a general workbench for analysing regulatory sequence regions and discovering transcription factor binding sites and cis-regulatory modules. MotifLab supports comprehensive motif discovery and analysis by allowing users to integrate several popular motif discovery tools as well as different kinds of additional information, including phylogenetic conservation, epigenetic marks, DNase hypersensitive sites, ChIP-Seq data, positional binding preferences of transcription factors, transcription factor interactions and gene expression. MotifLab offers several data-processing operations that can be used to create, manipulate and analyse data objects, and complete analysis workflows can be constructed and automatically executed within MotifLab, including graphical presentation of the results.
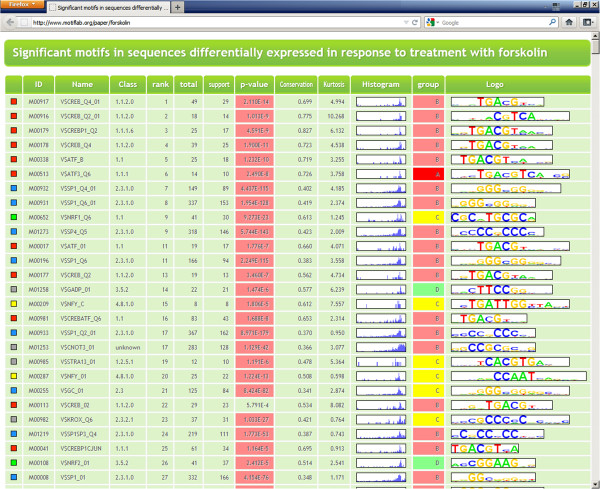
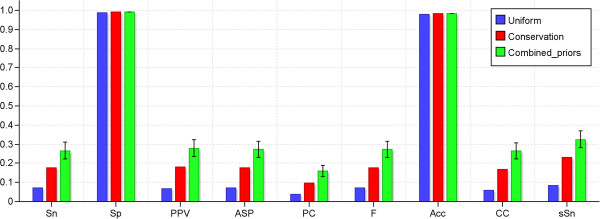
We have developed MotifLab as a flexible workbench for motif analysis in a genomic context. The flexibility and effectiveness of this workbench has been demonstrated on selected test cases, in particular two previously published benchmark data sets for single motifs and modules, and a realistic example of genes responding to treatment with forskolin. MotifLab is freely available at http://www.motiflab.org.
传统的计算基序发现方法通常性能不佳。特别是,搜索与已知结合基序序列匹配的方法往往会预测许多非功能结合位点,因为它们没有考虑到细胞的生物学状态。近年来,全基因组研究产生了大量的数据,这些数据有可能提高我们识别功能基序和结合位点的能力,例如不同细胞类型中染色质可及性和表观遗传状态的信息。然而,将这些数据与现有的基序发现工具结合使用并不总是一件简单的事情,特别是对于不擅长生物信息学编程的研究人员来说。
在这里,我们提出了 MotifLab,这是一个用于分析调控序列区域和发现转录因子结合位点和顺式调控模块的通用工作台。MotifLab 通过允许用户集成几个流行的基序发现工具以及不同类型的附加信息,包括系统发育保守性、表观遗传标记、DNase 超敏位点、ChIP-Seq 数据、转录因子的位置结合偏好、转录因子相互作用和基因表达,来支持全面的基序发现和分析。MotifLab 提供了几种数据处理操作,可以用于创建、操作和分析数据对象,并且可以在 MotifLab 内构建和自动执行完整的分析工作流程,包括结果的图形表示。
我们已经开发了 MotifLab 作为基因组背景下基序分析的灵活工作台。该工作台的灵活性和有效性已在选定的测试用例中得到证明,特别是两个以前发表的用于单基序和模块的基准数据集,以及一个使用 forskolin 处理基因的现实示例。MotifLab 可在 http://www.motiflab.org 免费获得。