Plant Research International, P.O. Box 6708 PB, Wageningen, The Netherlands.
BMC Genomics. 2013 Jan 16;14:21. doi: 10.1186/1471-2164-14-21.
The genome of Fusarium graminearum has been sequenced and annotated previously, but correct gene annotation remains a challenge. In addition, posttranscriptional regulations, such as alternative splicing and RNA editing, are poorly understood in F. graminearum. Here we took advantage of RNA-Seq to improve gene annotations and to identify alternative splicing and RNA editing in F. graminearum.
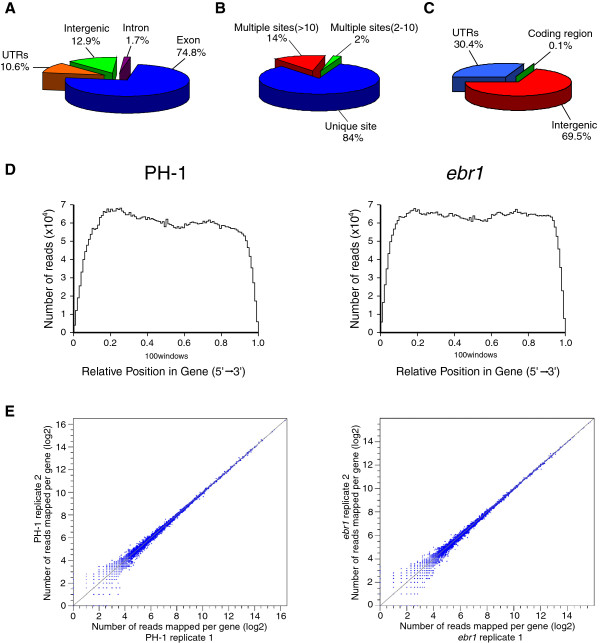
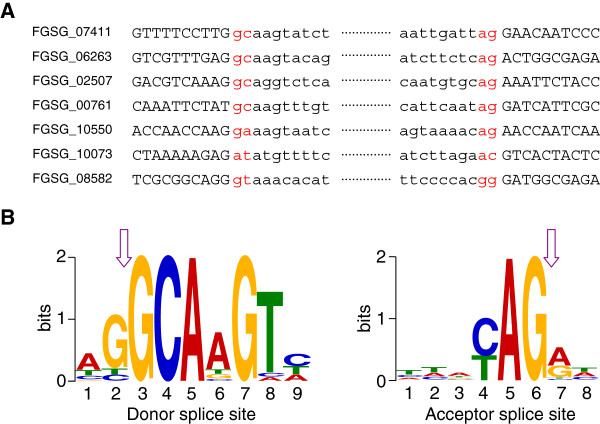
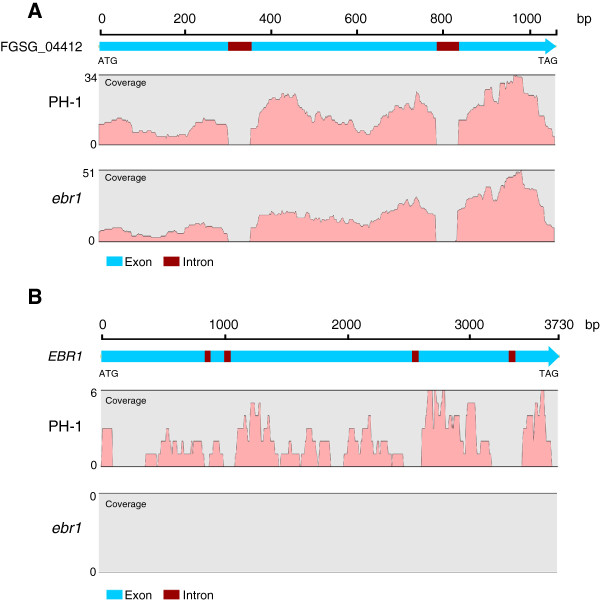
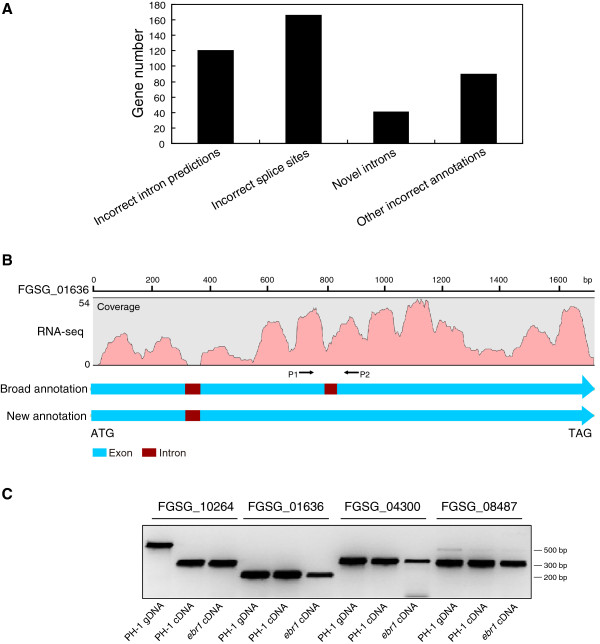
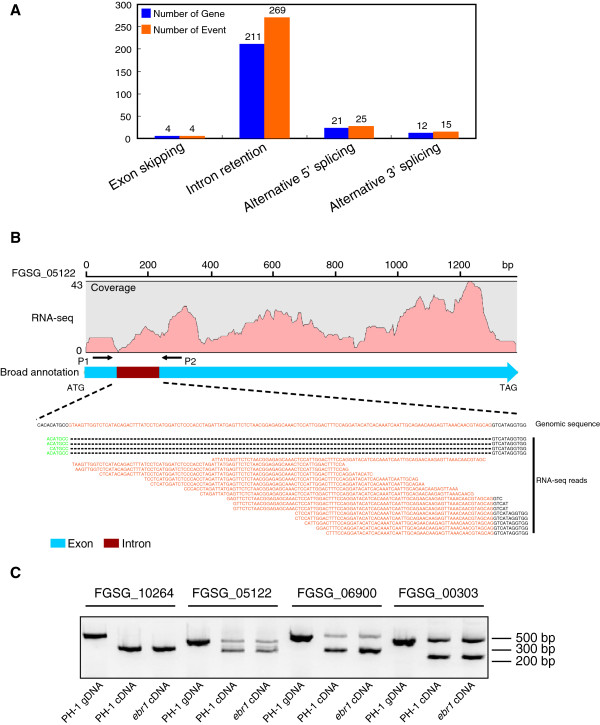
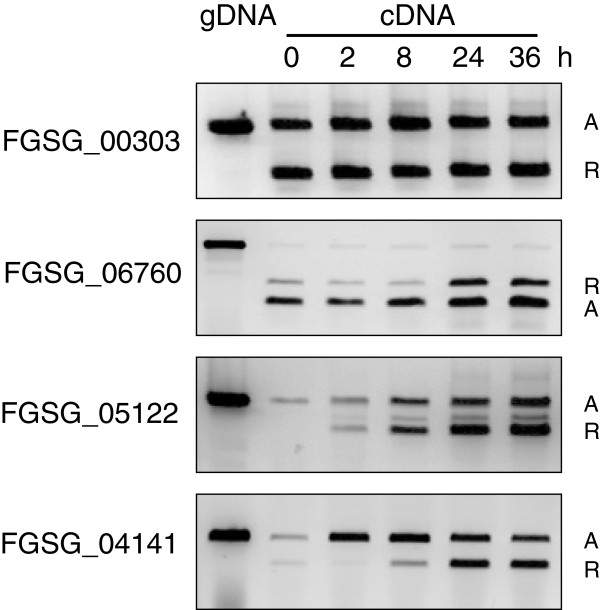
We identified and revised 655 incorrectly predicted gene models, including revisions of intron predictions, intron splice sites and prediction of novel introns. 231 genes were identified with two or more alternative splice variants, mostly due to intron retention. Interestingly, the expression ratios between different transcript isoforms appeared to be developmentally regulated. Surprisingly, no RNA editing was identified in F. graminearum. Moreover, 2459 novel transcriptionally active regions (nTARs) were identified and our analysis indicates that many of these could be missed genes. Finally, we identified the 5' UTR and/or 3' UTR sequences of 7666 genes. A number of representative novel gene models and alternatively spliced genes were validated by reverse transcription polymerase chain reaction and sequencing of the generated amplicons.
We have developed novel and efficient strategies to identify alternatively spliced genes and incorrect gene models based on RNA-Seq data. Our study identified hundreds of alternatively spliced genes in F. graminearum and for the first time indicated that alternative splicing is developmentally regulated in filamentous fungi. In addition, hundreds of incorrect predicted gene models were identified and revised and thousands of nTARs were discovered in our study, which will be helpful for the future genomic and transcriptomic studies in F. graminearum.
禾谷镰刀菌的基因组已经测序并注释,但正确的基因注释仍然是一个挑战。此外,禾谷镰刀菌中转录后调控,如选择性剪接和 RNA 编辑,知之甚少。在这里,我们利用 RNA-Seq 来改进基因注释,并鉴定禾谷镰刀菌中的选择性剪接和 RNA 编辑。
我们鉴定并修正了 655 个错误预测的基因模型,包括内含子预测、内含子剪接位点和新内含子的预测修正。鉴定出 231 个基因具有两个或更多的选择性剪接变体,主要是由于内含子保留。有趣的是,不同转录本异构体之间的表达比例似乎受到发育调控。令人惊讶的是,禾谷镰刀菌中没有发现 RNA 编辑。此外,鉴定出 2459 个新的转录活性区(nTAR),我们的分析表明,其中许多可能是缺失基因。最后,我们鉴定了 7666 个基因的 5'UTR 和/或 3'UTR 序列。许多代表性的新基因模型和选择性剪接基因通过逆转录聚合酶链反应和生成的扩增子测序得到验证。
我们开发了基于 RNA-Seq 数据鉴定选择性剪接基因和错误基因模型的新方法。我们的研究在禾谷镰刀菌中鉴定了数百个选择性剪接基因,首次表明选择性剪接在丝状真菌中受到发育调控。此外,在我们的研究中还鉴定和修正了数百个错误预测的基因模型,并发现了数千个 nTAR,这将有助于未来对禾谷镰刀菌的基因组和转录组研究。