Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK.
Chembiochem. 2013 Feb 11;14(3):332-42. doi: 10.1002/cbic.201200521. Epub 2013 Jan 23.
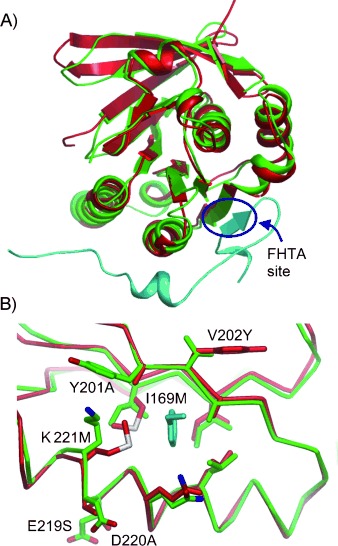
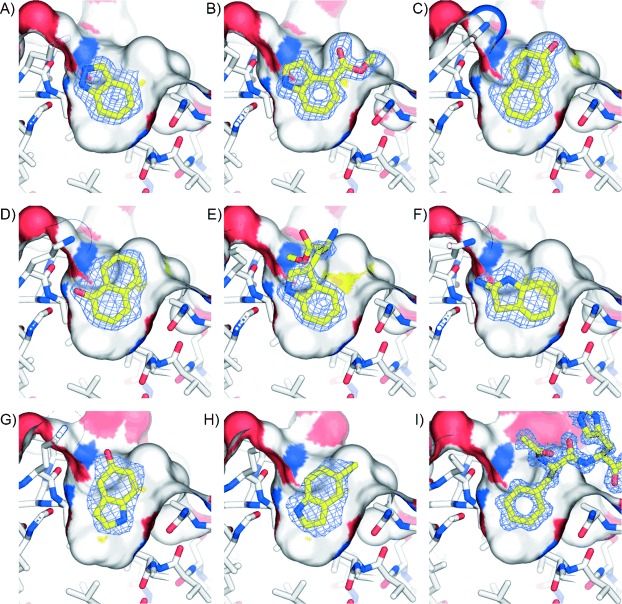
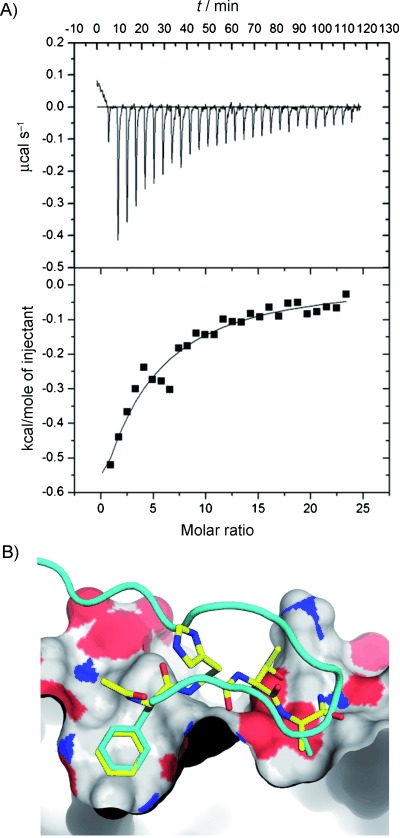
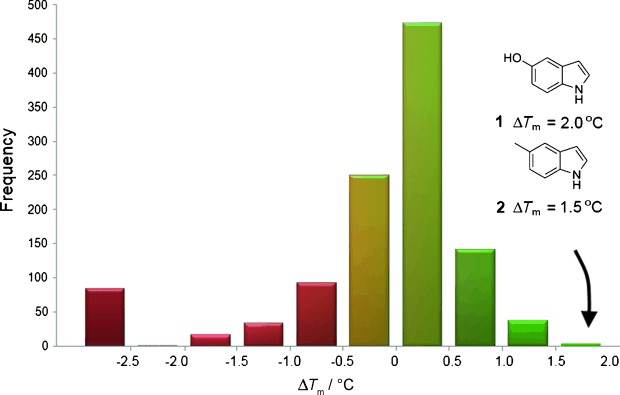
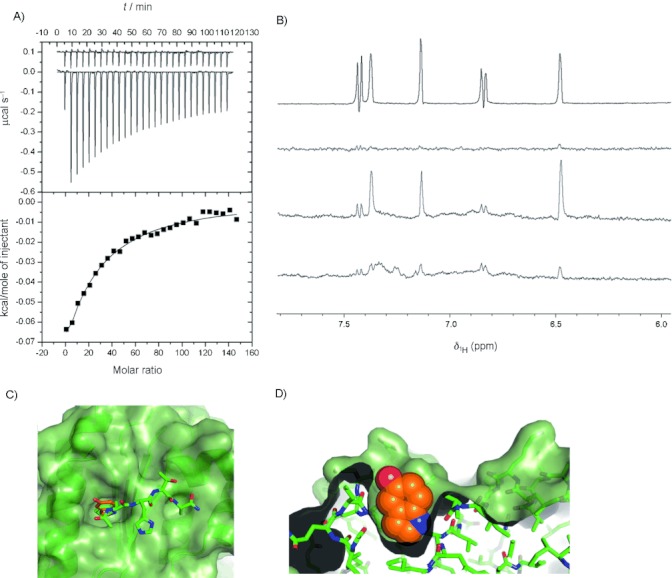
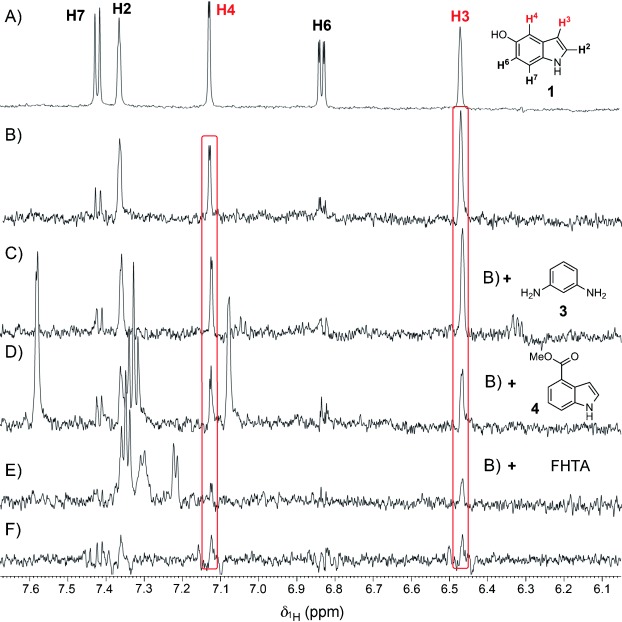
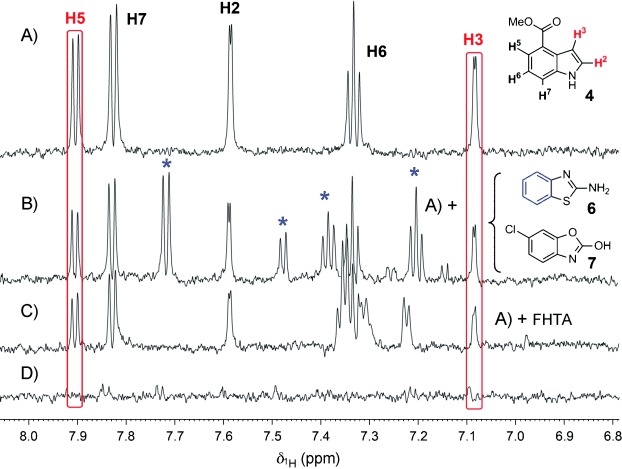
The ability to identify inhibitors of protein-protein interactions represents a major challenge in modern drug discovery and in the development of tools for chemical biology. In recent years, fragment-based approaches have emerged as a new methodology in drug discovery; however, few examples of small molecules that are active against chemotherapeutic targets have been published. Herein, we describe the fragment-based approach of targeting the interaction between the tumour suppressor BRCA2 and the recombination enzyme RAD51; it makes use of a screening pipeline of biophysical techniques that we expect to be more generally applicable to similar targets. Disruption of this interaction in vivo is hypothesised to give rise to cellular hypersensitivity to radiation and genotoxic drugs. We have used protein engineering to create a monomeric form of RAD51 by humanising a thermostable archaeal orthologue, RadA, and used this protein for fragment screening. The initial fragment hits were thoroughly validated biophysically by isothermal titration calorimetry (ITC) and NMR techniques and observed by X-ray crystallography to bind in a shallow surface pocket that is occupied in the native complex by the side chain of a phenylalanine from the conserved FxxA interaction motif found in BRCA2. This represents the first report of fragments or any small molecule binding at this protein-protein interaction site.
鉴定蛋白质-蛋白质相互作用抑制剂是现代药物发现和化学生物学工具开发的主要挑战。近年来,基于片段的方法已成为药物发现的新方法;然而,很少有针对化学治疗靶点的小分子具有活性的例子被发表。在此,我们描述了针对肿瘤抑制因子 BRCA2 与重组酶 RAD51 之间相互作用的基于片段的方法;它利用了我们预期更普遍适用于类似靶标的生物物理筛选技术。假设破坏这种相互作用会导致细胞对辐射和遗传毒性药物的敏感性增加。我们使用蛋白质工程通过人源化耐热古细菌同源物 RadA 来创建 RAD51 的单体形式,并将该蛋白质用于片段筛选。最初的片段命中物通过等温滴定量热法(ITC)和 NMR 技术进行了全面的生物物理验证,并通过 X 射线晶体学观察到结合在一个浅表面口袋中,该口袋在天然复合物中被来自 BRCA2 中保守 FxxA 相互作用基序的苯丙氨酸侧链占据。这代表了第一个报告片段或任何小分子结合在该蛋白质-蛋白质相互作用位点的报告。