Mahidol-Oxford Tropical Medicine Research Unit, Faculty of Tropical Medicine, Mahidol University, Bangkok, Thailand.
PLoS Negl Trop Dis. 2013;7(1):e1954. doi: 10.1371/journal.pntd.0001954. Epub 2013 Jan 24.
The available Leptospira multilocus sequence typing (MLST) scheme supported by a MLST website is limited to L. interrogans and L. kirschneri. Our aim was to broaden the utility of this scheme to incorporate a total of seven pathogenic species.
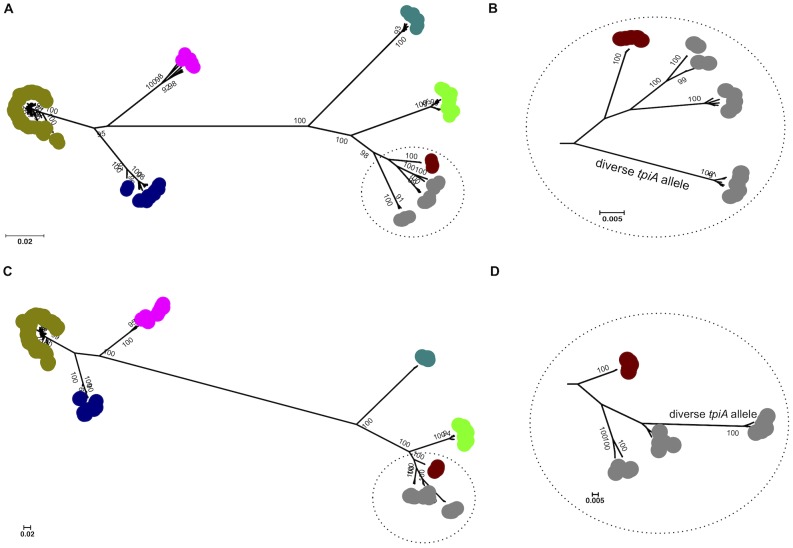
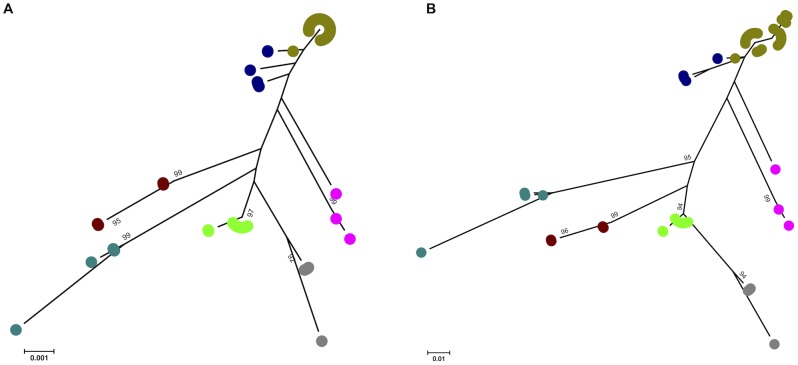
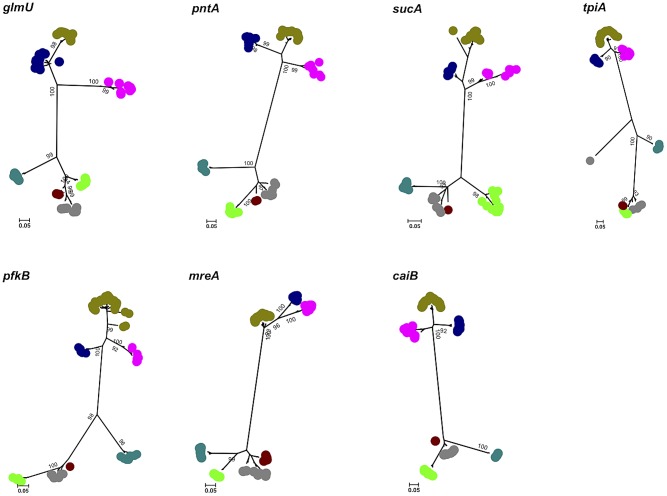
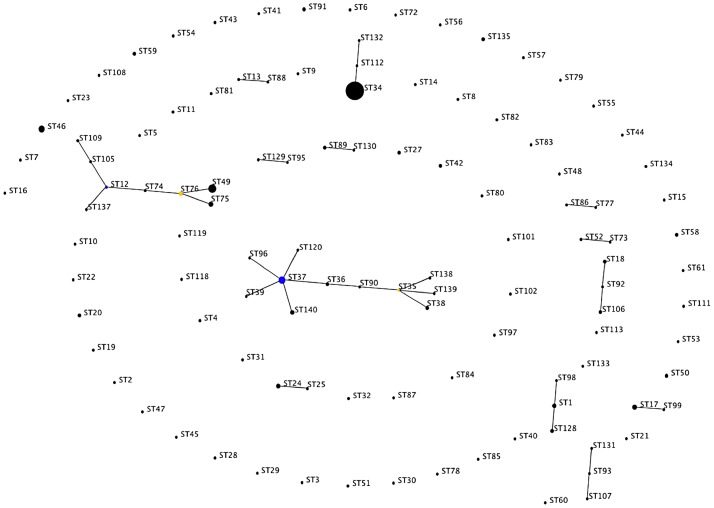
We modified the existing scheme by replacing one of the seven MLST loci (fadD was changed to caiB), as the former gene did not appear to be present in some pathogenic species. Comparison of the original and modified schemes using data for L. interrogans and L. kirschneri demonstrated that the discriminatory power of the two schemes was not significantly different. The modified scheme was used to further characterize 325 isolates (L. alexanderi [n = 5], L. borgpetersenii [n = 34], L. interrogans [n = 222], L. kirschneri [n = 29], L. noguchii [n = 9], L. santarosai [n = 10], and L. weilii [n = 16]). Phylogenetic analysis using concatenated sequences of the 7 loci demonstrated that each species corresponded to a discrete clade, and that no strains were misclassified at the species level. Comparison between genotype and serovar was possible for 254 isolates. Of the 31 sequence types (STs) represented by at least two isolates, 18 STs included isolates assigned to two or three different serovars. Conversely, 14 serovars were identified that contained between 2 to 10 different STs. New observations were made on the global phylogeography of Leptospira spp., and the utility of MLST in making associations between human disease and specific maintenance hosts was demonstrated.
The new MLST scheme, supported by an updated MLST website, allows the characterization and species assignment of isolates of the seven major pathogenic species associated with leptospirosis.
现有的钩端螺旋体多位点序列分型(MLST)方案在一个 MLST 网站上支持的仅限于问号钩端螺旋体和克里夫兰钩端螺旋体。我们的目的是扩大该方案的应用范围,纳入总共七种致病性种。
我们通过替换七个 MLST 基因座中的一个(将 fadD 替换为 caiB)来修改现有的方案,因为前者基因似乎不存在于某些致病性种中。使用问号钩端螺旋体和克里夫兰钩端螺旋体的数据比较原始方案和修改方案,证明两个方案的区分力没有显著差异。使用修改后的方案进一步对 325 株分离株进行了特征描述(亚历山大钩端螺旋体[n=5],波尔图钩端螺旋体[n=34],问号钩端螺旋体[n=222],克里夫兰钩端螺旋体[n=29],努果茨钩端螺旋体[n=9],圣罗莎钩端螺旋体[n=10],和维尔钩端螺旋体[n=16])。使用 7 个基因座的串联序列进行的系统发育分析表明,每个种都对应一个离散的分支,在种水平上没有菌株被错误分类。对 254 株分离株的基因型和血清型进行了比较。至少有两个分离株代表的 31 个序列型(ST)中,18 个 ST 包括被分配到两个或三个不同血清型的分离株。相反,鉴定出 14 个血清型,其中包含 2 到 10 个不同的 ST。在钩端螺旋体种的全球系统地理学方面有了新的发现,并证明了 MLST 在将人类疾病与特定维持宿主联系起来的应用。
新的 MLST 方案在一个更新的 MLST 网站上得到支持,允许对与钩端螺旋体病相关的七种主要致病性种的分离株进行特征描述和种属分配。