Rahman Atif, Pachter Lior
Genome Biol. 2013 Jan 29;14(1):R8. doi: 10.1186/gb-2013-14-1-r8.
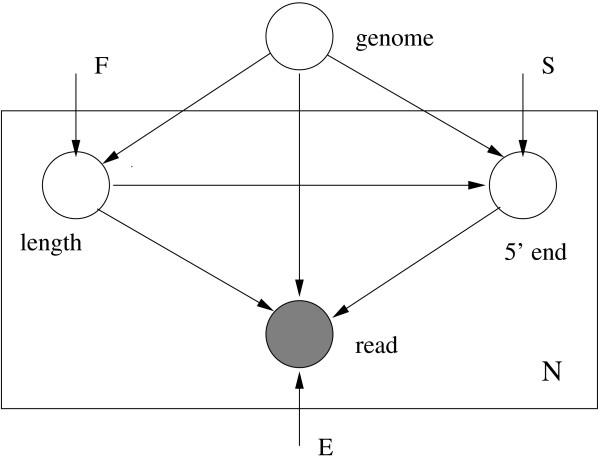
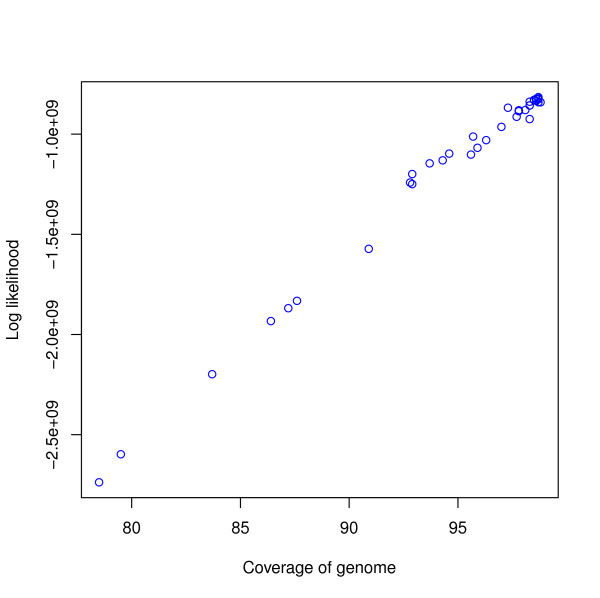
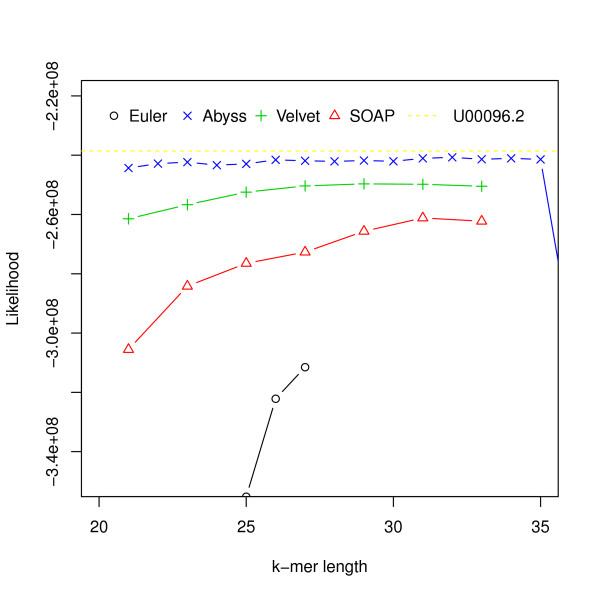
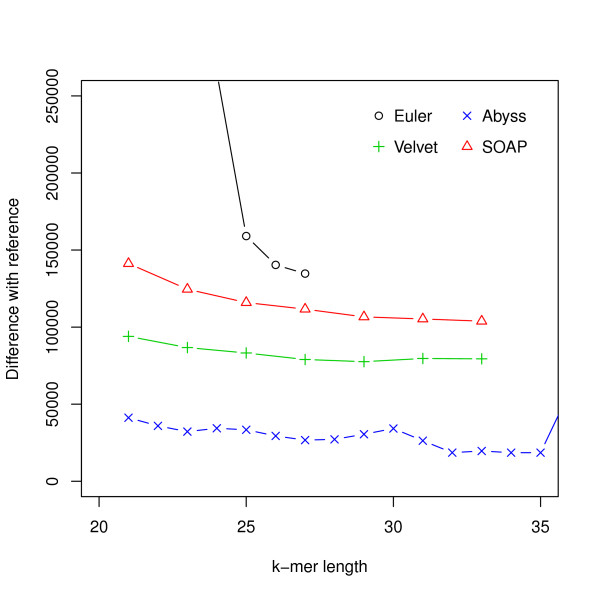
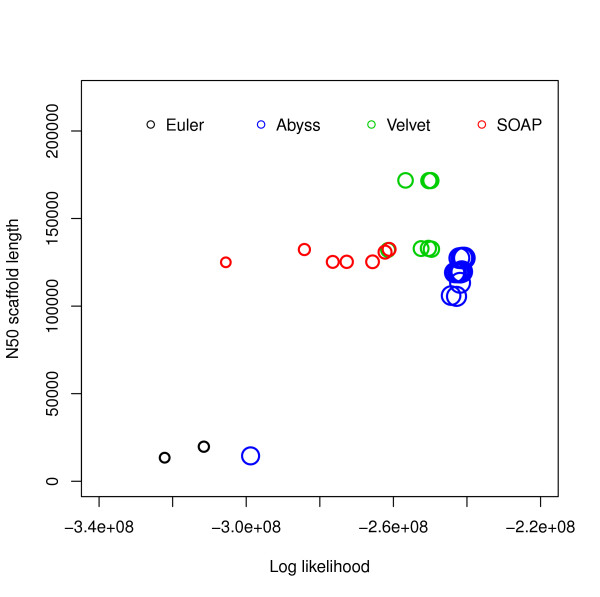
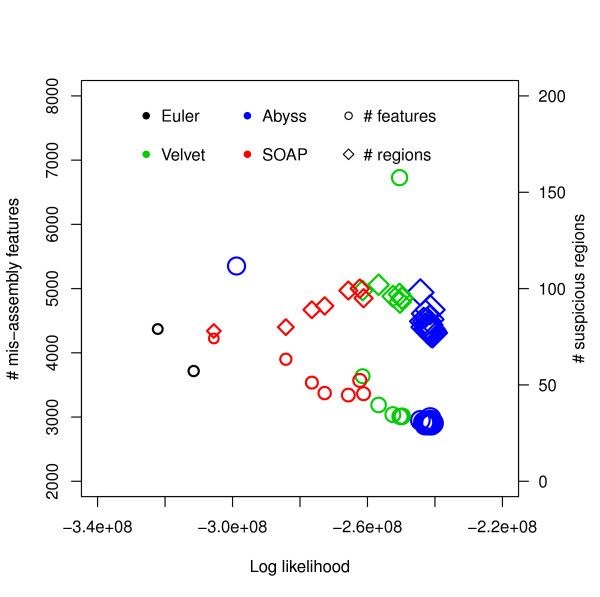
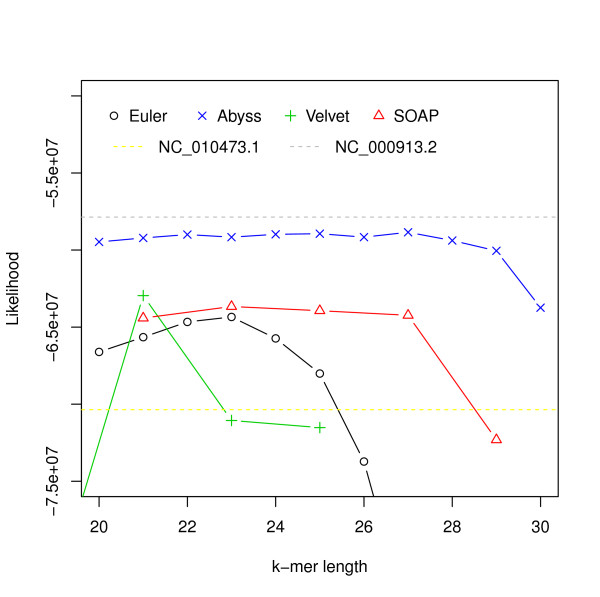
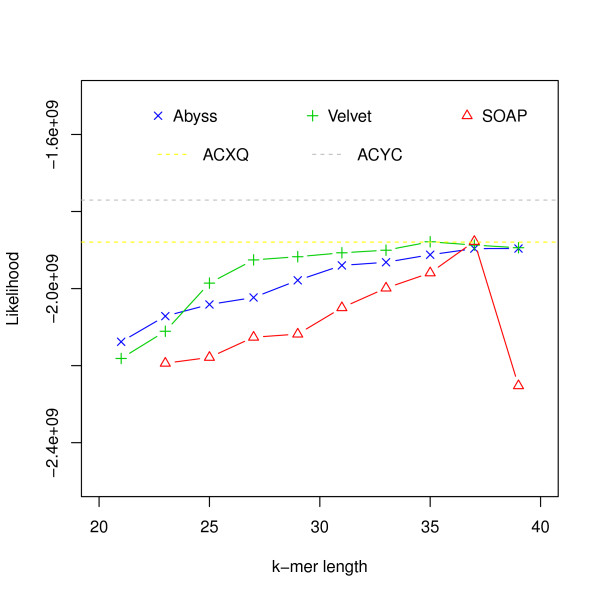
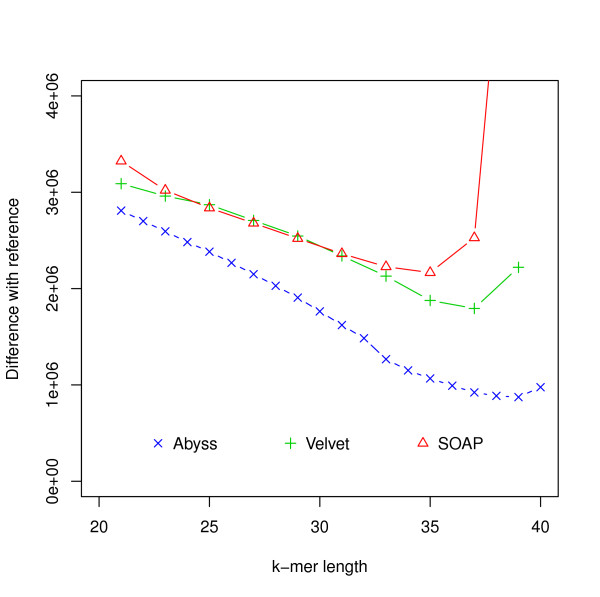
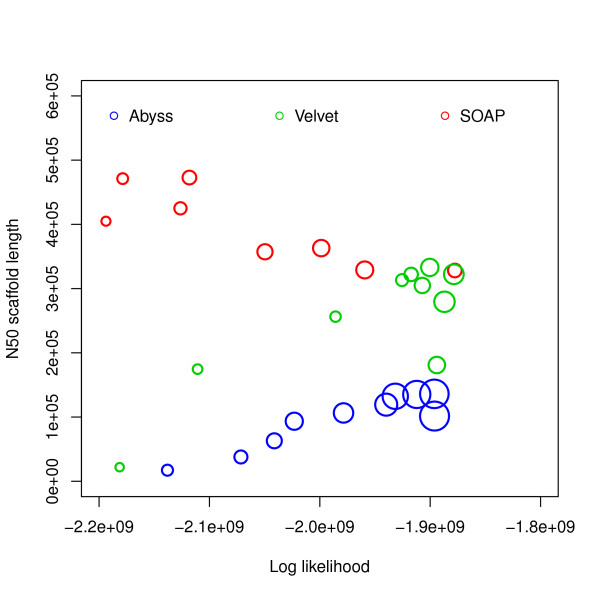
Assembly algorithms have been extensively benchmarked using simulated data so that results can be compared to ground truth. However, in de novo assembly, only crude metrics such as contig number and size are typically used to evaluate assembly quality. We present CGAL, a novel likelihood-based approach to assembly assessment in the absence of a ground truth. We show that likelihood is more accurate than other metrics currently used for evaluating assemblies, and describe its application to the optimization and comparison of assembly algorithms. Our methods are implemented in software that is freely available at http://bio.math.berkeley.edu/cgal/.
组装算法已经使用模拟数据进行了广泛的基准测试,以便将结果与真实情况进行比较。然而,在从头组装中,通常仅使用诸如重叠群数量和大小等粗略指标来评估组装质量。我们提出了CGAL,这是一种在没有真实情况的情况下进行组装评估的基于似然性的新方法。我们表明,似然性比目前用于评估组装的其他指标更准确,并描述了其在组装算法优化和比较中的应用。我们的方法在软件中实现,该软件可在http://bio.math.berkeley.edu/cgal/免费获取。