Institute for Genetics and Biometry, Department of Bioinformatics and Biomathematics, Leibniz Institute for Farm Animal Biology, Dummerstorf, Germany.
PLoS One. 2013;8(2):e55267. doi: 10.1371/journal.pone.0055267. Epub 2013 Feb 11.
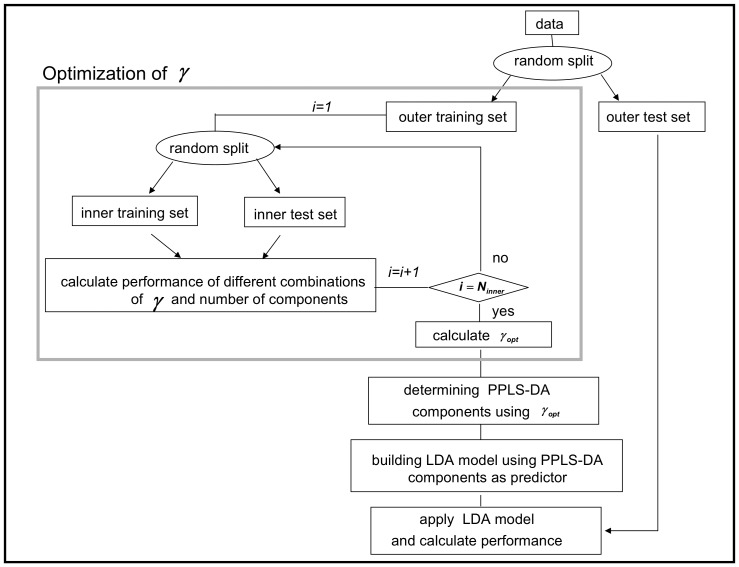
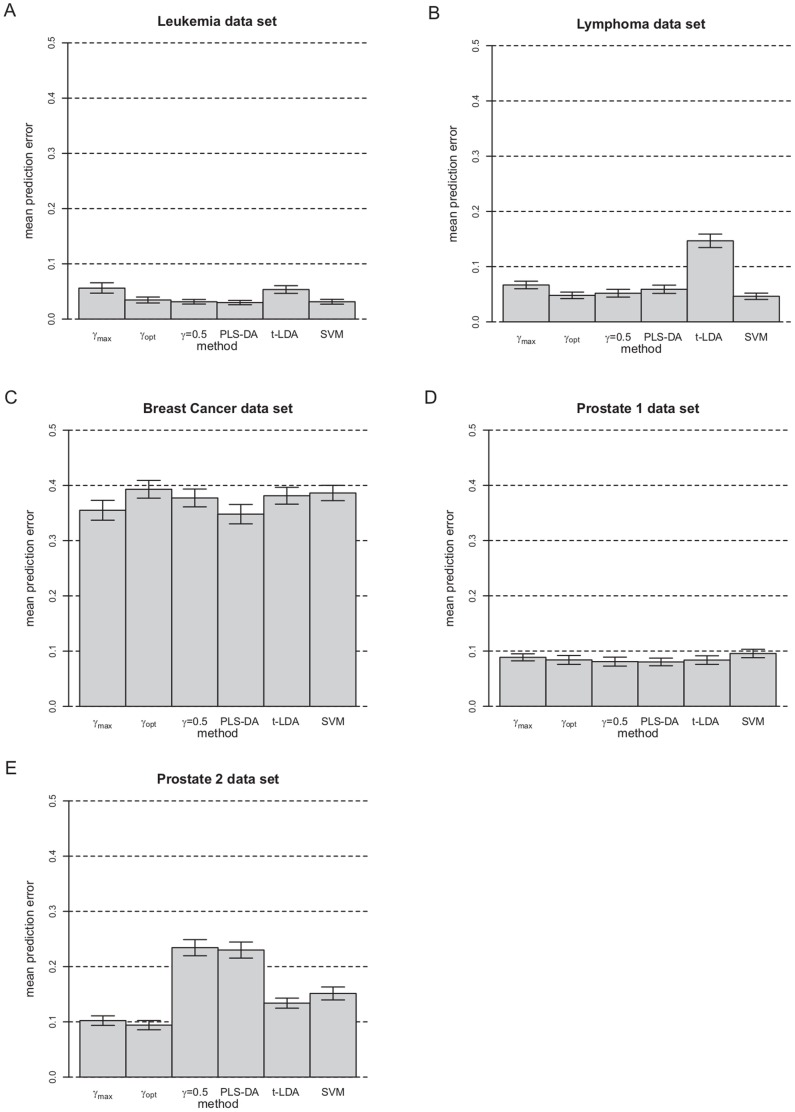
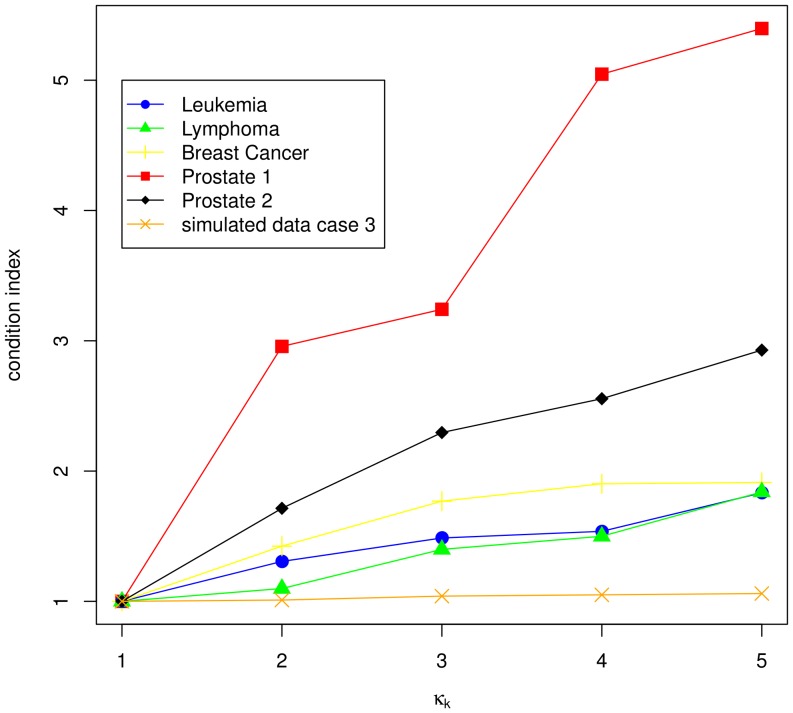
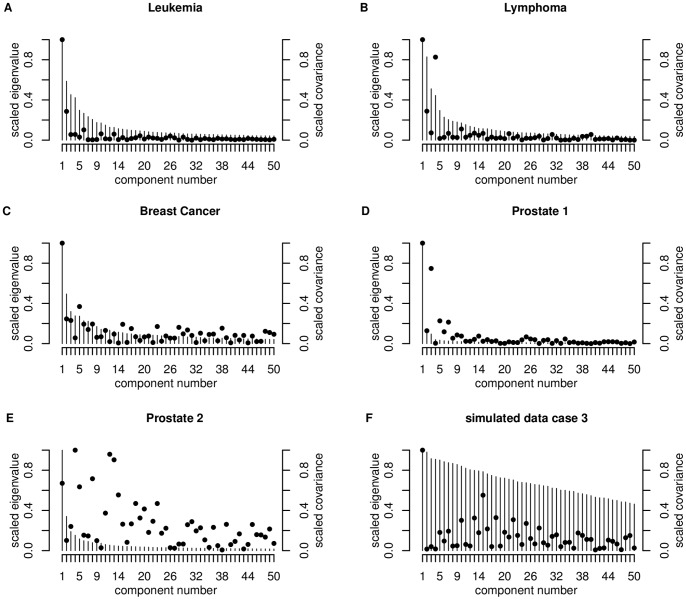
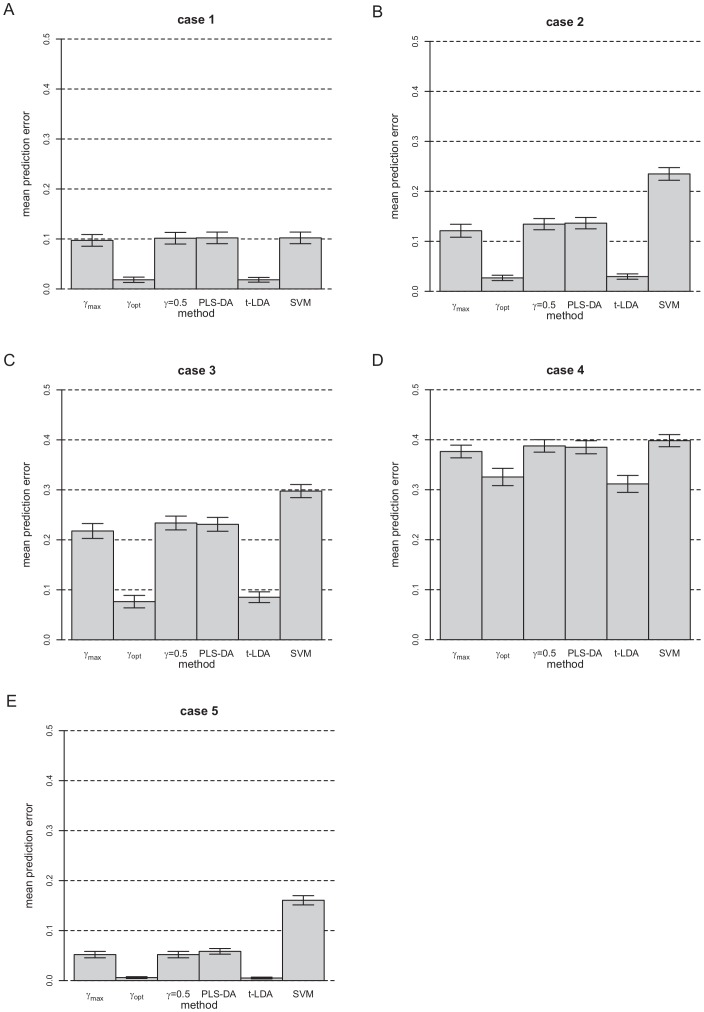
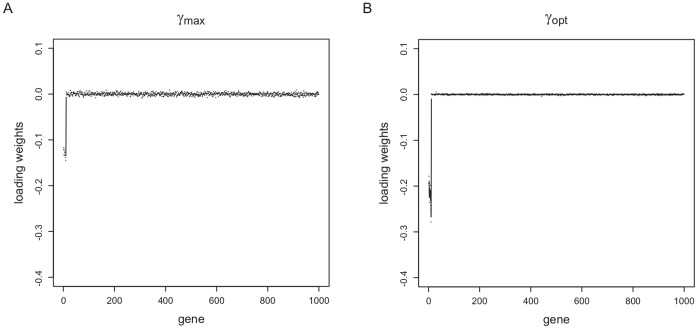
Classification studies are widely applied, e.g. in biomedical research to classify objects/patients into predefined groups. The goal is to find a classification function/rule which assigns each object/patient to a unique group with the greatest possible accuracy (classification error). Especially in gene expression experiments often a lot of variables (genes) are measured for only few objects/patients. A suitable approach is the well-known method PLS-DA, which searches for a transformation to a lower dimensional space. Resulting new components are linear combinations of the original variables. An advancement of PLS-DA leads to PPLS-DA, introducing a so called 'power parameter', which is maximized towards the correlation between the components and the group-membership. We introduce an extension of PPLS-DA for optimizing this power parameter towards the final aim, namely towards a minimal classification error. We compare this new extension with the original PPLS-DA and also with the ordinary PLS-DA using simulated and experimental datasets. For the investigated data sets with weak linear dependency between features/variables, no improvement is shown for PPLS-DA and for the extensions compared to PLS-DA. A very weak linear dependency, a low proportion of differentially expressed genes for simulated data, does not lead to an improvement of PPLS-DA over PLS-DA, but our extension shows a lower prediction error. On the contrary, for the data set with strong between-feature collinearity and a low proportion of differentially expressed genes and a large total number of genes, the prediction error of PPLS-DA and the extensions is clearly lower than for PLS-DA. Moreover we compare these prediction results with results of support vector machines with linear kernel and linear discriminant analysis.
分类研究被广泛应用,例如在生物医学研究中,将对象/患者分类到预定义的组中。目标是找到一个分类函数/规则,将每个对象/患者分配到一个具有最大可能准确性(分类误差)的唯一组中。特别是在基因表达实验中,通常为少数对象/患者测量了大量变量(基因)。一种合适的方法是众所周知的 PLS-DA 方法,它搜索到一个较低维数的空间的转换。产生的新组件是原始变量的线性组合。PLS-DA 的一个改进是 PPLS-DA,引入了所谓的“幂参数”,该参数在组件和组成员之间的相关性方面最大化。我们引入了 PPLS-DA 的扩展,以优化该幂参数以实现最终目标,即最小化分类误差。我们将这种新扩展与原始 PPLS-DA 进行比较,也与使用模拟和实验数据集的普通 PLS-DA 进行比较。对于所研究的具有特征/变量之间弱线性相关性的数据集,与 PLS-DA 相比,PPLS-DA 和扩展没有显示出改进。对于模拟数据,特征之间的弱线性相关性和差异表达基因的低比例,不会导致 PLS-DA 优于 PPLS-DA,但我们的扩展显示出更低的预测误差。相反,对于具有强特征之间共线性和低比例差异表达基因和大量基因的数据集,PPLS-DA 和扩展的预测误差明显低于 PLS-DA。此外,我们将这些预测结果与具有线性核和线性判别分析的支持向量机的结果进行了比较。