Hicks Chindo, Kumar Ranjit, Pannuti Antonio, Backus Kandis, Brown Alexandra, Monico Jesus, Miele Lucio
Cancer Institute, University of Mississippi Medical Center, Jackson, MS. ; Department of Medicine, University of Mississippi Medical Center, Jackson, MS.
Cancer Inform. 2013;12:1-20. doi: 10.4137/CIN.S10413. Epub 2013 Jan 29.
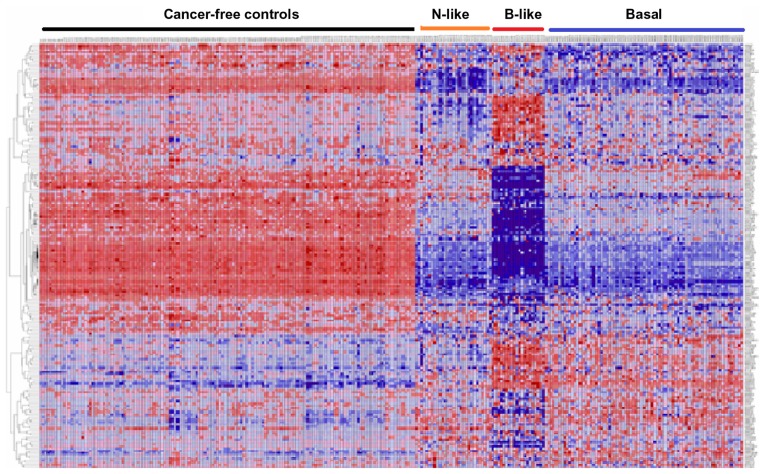
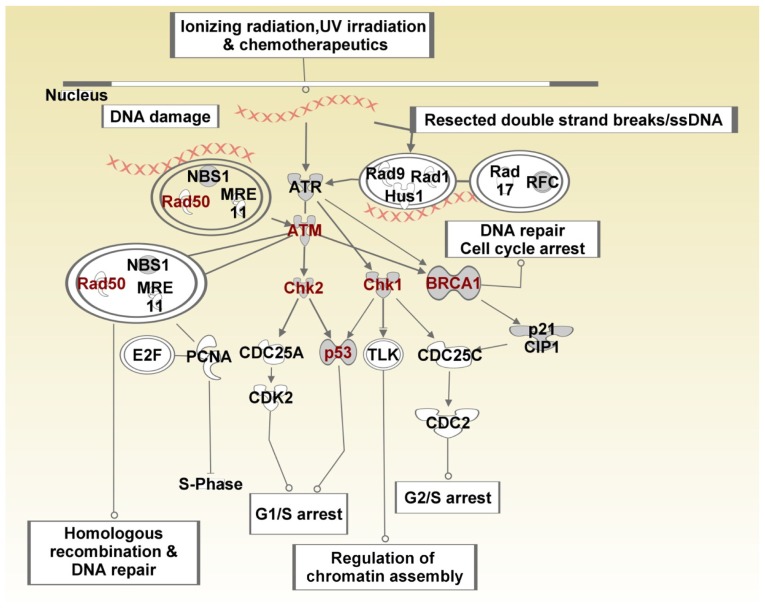
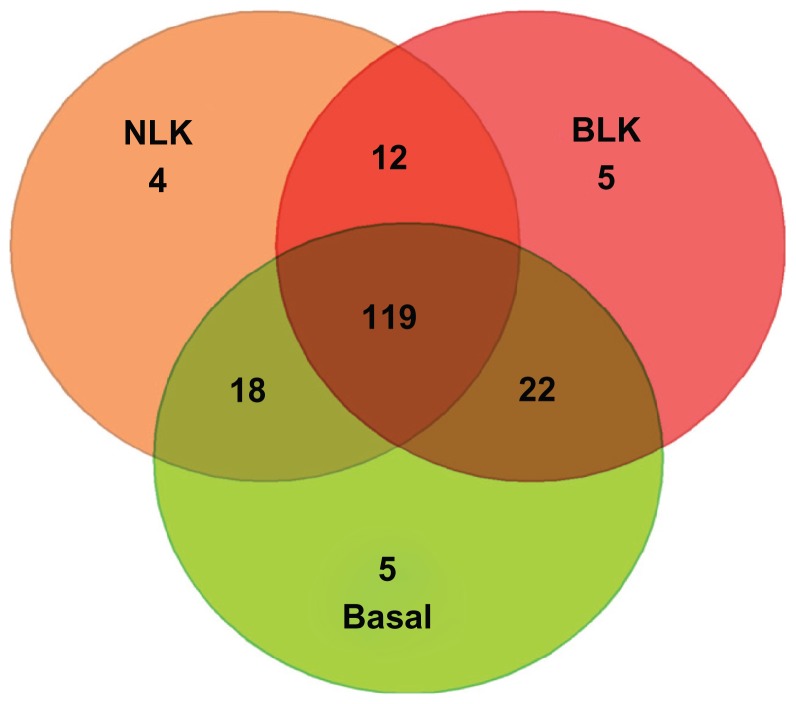
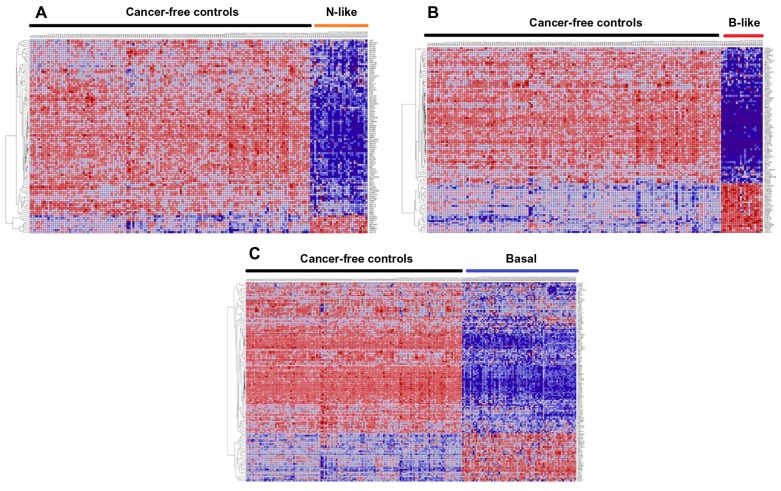
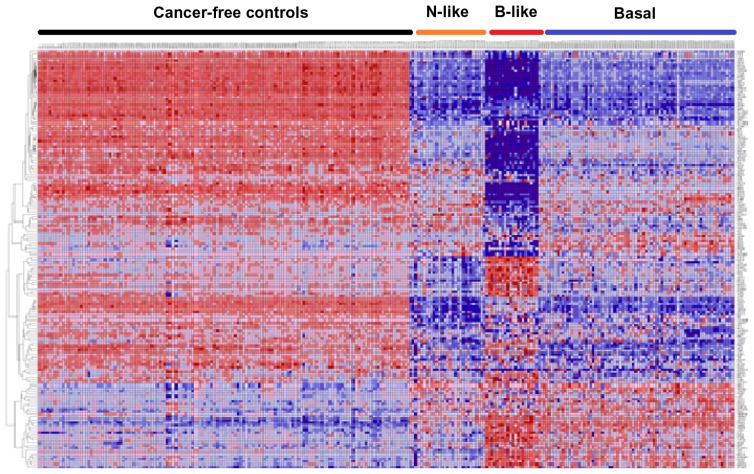
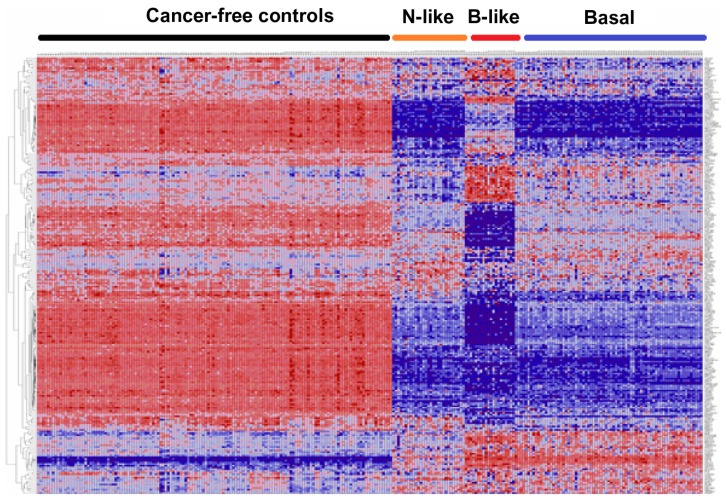
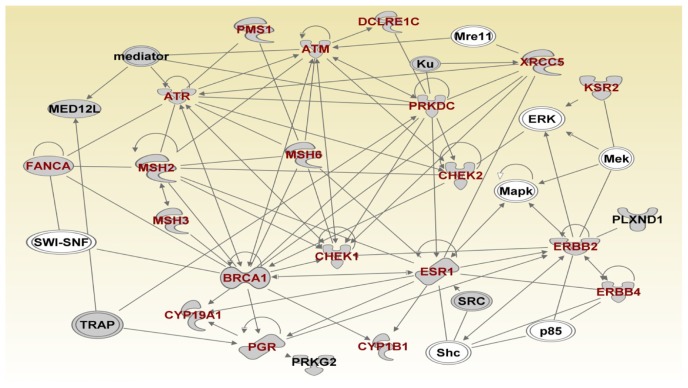
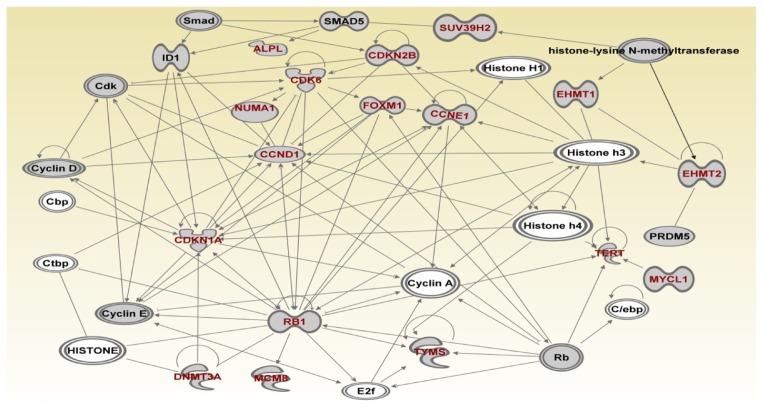
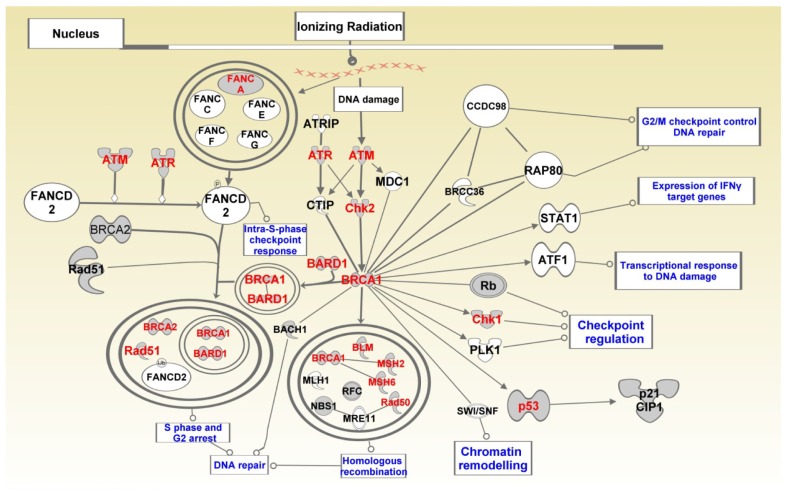
Genome-wide association studies (GWAS) have identified genetic variants associated with an increased risk of developing breast cancer. However, the association of genetic variants and their associated genes with the most aggressive subset of breast cancer, the triple-negative breast cancer (TNBC), remains a central puzzle in molecular epidemiology. The objective of this study was to determine whether genes containing single nucleotide polymorphisms (SNPs) associated with an increased risk of developing breast cancer are connected to and could stratify different subtypes of TNBC. Additionally, we sought to identify molecular pathways and networks involved in TNBC. We performed integrative genomics analysis, combining information from GWAS studies involving over 400,000 cases and over 400,000 controls, with gene expression data derived from 124 breast cancer patients classified as TNBC (at the time of diagnosis) and 142 cancer-free controls. Analysis of GWAS reports produced 500 SNPs mapped to 188 genes. We identified a signature of 159 functionally related SNP-containing genes which were significantly (P <10(-5)) associated with and stratified TNBC. Additionally, we identified 97 genes which were functionally related to, and had similar patterns of expression profiles, SNP-containing genes. Network modeling and pathway prediction revealed multi-gene pathways including p53, NFkB, BRCA, apoptosis, DNA repair, DNA mismatch, and excision repair pathways enriched for SNPs mapped to genes significantly associated with TNBC. The results provide convincing evidence that integrating GWAS information with gene expression data provides a unified and powerful approach for biomarker discovery in TNBC.
全基因组关联研究(GWAS)已经确定了与患乳腺癌风险增加相关的基因变异。然而,基因变异及其相关基因与乳腺癌最具侵袭性的亚型——三阴性乳腺癌(TNBC)之间的关联,仍然是分子流行病学中的一个核心难题。本研究的目的是确定含有与患乳腺癌风险增加相关的单核苷酸多态性(SNP)的基因是否与TNBC的不同亚型相关联并能够对其进行分层。此外,我们试图识别参与TNBC的分子途径和网络。我们进行了整合基因组分析,将来自涉及超过40万例病例和超过40万例对照的GWAS研究的信息,与来自124例被分类为TNBC(诊断时)的乳腺癌患者和142例无癌对照的基因表达数据相结合。对GWAS报告的分析产生了500个映射到188个基因的SNP。我们确定了159个功能相关的含SNP基因的特征,这些基因与TNBC显著相关(P <10^(-5))并对其进行了分层。此外,我们还确定了97个与含SNP基因功能相关且具有相似表达谱模式的基因。网络建模和途径预测揭示了多基因途径,包括p53、NFkB、BRCA、细胞凋亡、DNA修复、DNA错配和切除修复途径,这些途径富含映射到与TNBC显著相关的基因的SNP。结果提供了令人信服的证据,表明将GWAS信息与基因表达数据相结合为TNBC中的生物标志物发现提供了一种统一而强大的方法。