Research Center for Functional Genomics, Biomedicine and Translational Medicine, Iuliu Hatieganu University of Medicine and Pharmacy, 400337 Cluj-Napoca, Cluj, Romania.
MEDFUTURE Research Center for Advanced Medicine, Iuliu Hatieganu University of Medicine and Pharmacy, 400337 Cluj-Napoca, Romania.
Int J Mol Sci. 2020 Aug 14;21(16):5835. doi: 10.3390/ijms21165835.
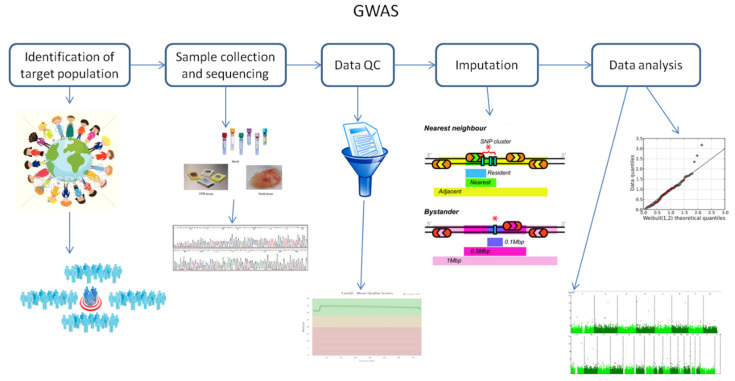
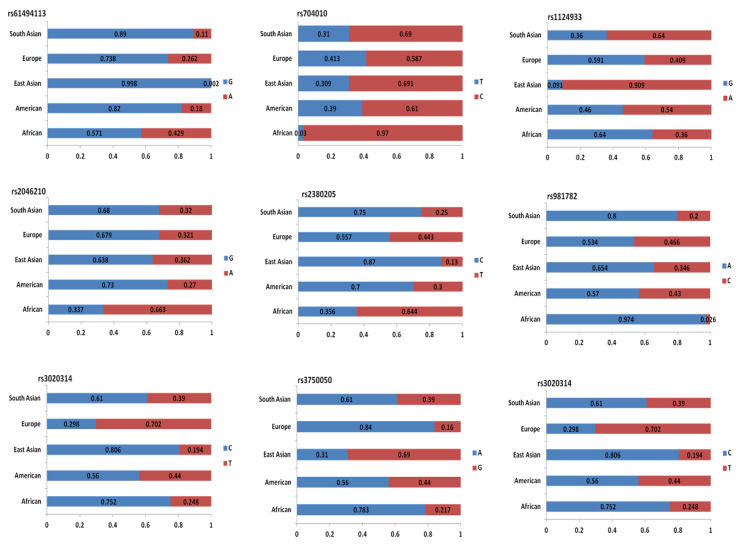
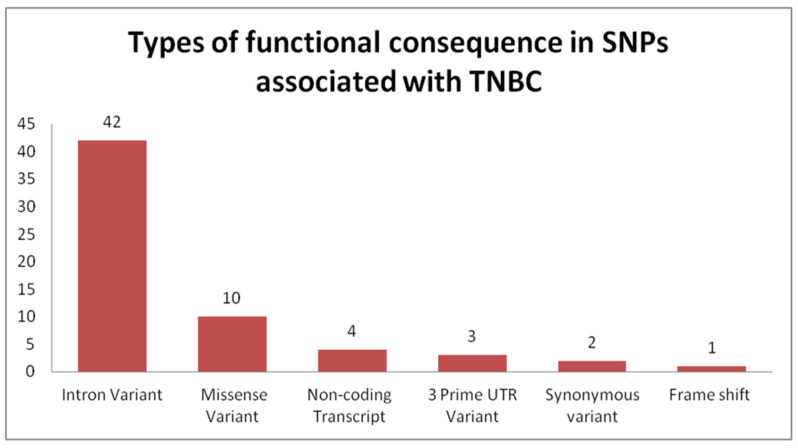
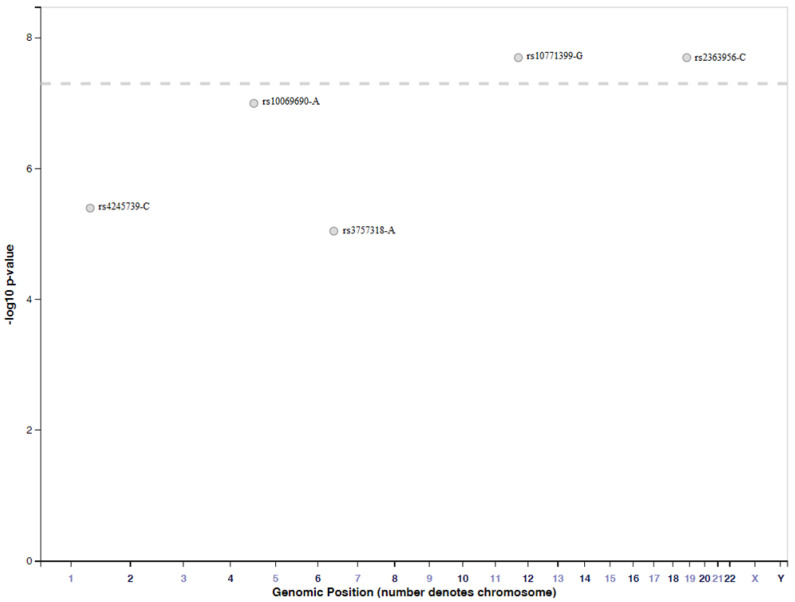
Genome-wide association studies (GWAS) are useful in assessing and analyzing either differences or variations in DNA sequences across the human genome to detect genetic risk factors of diseases prevalent within a target population under study. The ultimate goal of GWAS is to predict either disease risk or disease progression by identifying genetic risk factors. These risk factors will define the biological basis of disease susceptibility for the purposes of developing innovative, preventative, and therapeutic strategies. As single nucleotide polymorphisms (SNPs) are often used in GWAS, their relevance for triple negative breast cancer (TNBC) will be assessed in this review. Furthermore, as there are different levels and patterns of linkage disequilibrium (LD) present within different human subpopulations, a plausible strategy to evaluate known SNPs associated with incidence of breast cancer in ethnically different patient cohorts will be presented and discussed. Additionally, a description of GWAS for TNBC will be presented, involving various identified SNPs correlated with miRNA sites to determine their efficacies on either prognosis or progression of TNBC in patients. Although GWAS have identified multiple common breast cancer susceptibility variants that individually would result in minor risks, it is their combined effects that would likely result in major risks. Thus, one approach to quantify synergistic effects of such common variants is to utilize polygenic risk scores. Therefore, studies utilizing predictive risk scores (PRSs) based on known breast cancer susceptibility SNPs will be evaluated. Such PRSs are potentially useful in improving stratification for screening, particularly when combining family history, other risk factors, and risk prediction models. In conclusion, although interpretation of the results from GWAS remains a challenge, the use of SNPs associated with TNBC may elucidate and better contextualize these studies.
全基因组关联研究(GWAS)在评估和分析人类基因组中 DNA 序列的差异或变异方面非常有用,可用于检测研究人群中常见疾病的遗传风险因素。GWAS 的最终目标是通过识别遗传风险因素来预测疾病风险或疾病进展。这些风险因素将为疾病易感性确定生物学基础,从而开发创新的、预防性的和治疗性策略。由于单核苷酸多态性(SNP)常用于 GWAS,因此将在本综述中评估其与三阴性乳腺癌(TNBC)的相关性。此外,由于不同的人类亚群中存在不同水平和模式的连锁不平衡(LD),因此将提出并讨论一种评估与不同种族患者乳腺癌发病相关的已知 SNP 的合理策略。此外,还将介绍 TNBC 的 GWAS 描述,涉及与 miRNA 位点相关的各种已识别 SNP,以确定它们对 TNBC 患者预后或进展的疗效。尽管 GWAS 已经确定了多个常见的乳腺癌易感变体,这些变体单独会导致较小的风险,但它们的综合效应可能导致较大的风险。因此,量化此类常见变体协同效应的一种方法是利用多基因风险评分。因此,将评估基于已知乳腺癌易感 SNP 的预测风险评分(PRS)的研究。这种 PRS 可能有助于改善筛查的分层,特别是在结合家族史、其他风险因素和风险预测模型时。总之,尽管对 GWAS 结果的解释仍然是一个挑战,但与 TNBC 相关的 SNP 的使用可能会阐明和更好地说明这些研究。